
Abstract
Background:
Graves' hyperthyroidism is induced by immunizing mice with adenovirus expressing the human thyrotropin (TSH)-receptor. Using families of recombinant-inbred mice, we previously discovered that genetic susceptibility to induced thyroid-stimulating antibodies and hyperthyroidism are linked to loci on different chromosomes, indicating a fundamental genetic difference in thyroid sensitivity to ligand stimulation. An approach to assess thyroid sensitivity involves challenging genetically diverse lines of mice with TSH and measuring the genotype/strain-specific increase in serum thyroxine (T4).
Methods:
We investigated genetic susceptibility and genetic control of T4 stimulation by 10 mU bovine TSH in female mice of the CXB, BXH, and AXB/BXA strain families, all previously studied for induced Graves' hyperthyroidism.
Results:
Before TSH injection, T4 levels must be suppressed by inhibiting endogenous TSH secretion. Three daily intraperitoneal L-triiodothyronine injections efficiently suppressed serum T4 in females of 50 of 51 recombinant inbred strains. T4 stimulation by TSH was more strongly linked in CXB and BXH sets, derived from parental strains with divergent T4 stimulation, than in AXB/BXA strains generated from parents with similar TSH-induced responses. Genetic loci linked to the acute TSH-induced T4 response (hours) were not the same as those linked to induced hyperthyroidism (which develops over months).
Conclusions:
Genetic susceptibility for thyroid sensitivity to TSH stimulation was distinct for three families of inbred mouse lines. These observations parallel the human situation with multiple genetic loci contributing to the same trait and different loci associated with the same trait in different ethnic groups. Of the genetic loci highlighted in mice, three overlap with, or are located up or downstream, of human TSH-controlling genes. Other studies show that human disease genes can be identified through cross-species gene mapping of evolutionary conserved processes. Consequently, our findings suggest that novel thyroid function genes may yet be revealed in humans.
Introduction
Our previous studies of genetic susceptibility to induced hyperthyroidism were performed using three families of recombinant inbred strains: CXB, BXH, and AXB/BXA sets. Recombinant inbred lines are generated by crossing two inbred parental strains to provide the first filial generation (F1), crossing F1 progeny to generate the second generation (F2); subsequently, by repeated brother–sister F2 matings for 20 generations or more, to establish a set of stable homozygous and isogenic lines (8). High-resolution genetic maps, available for four families of recombinant inbred strains that share one parental strain (C57BL/6) provide powerful tools for mapping chromosomal loci linked with selected phenotypic traits [for example (9,10)].
An approach to assess thyroid sensitivity involves challenging different mouse lines with a defined dose of thyrotropin (TSH) and measuring the subsequent increase in serum thyroxine (T4). However, before TSH injection, it is necessary to rest the thyroid and suppress serum T4 levels by inhibiting endogenous TSH secretion. We previously have also shown that, unlike oral administration that is only effective in outbred or male mice, L-triiodothyronine (L-T3) injected intraperitoneally (i.p.) for 3 days efficiently suppresses females belonging to different inbred strains (11).
In the present study, we investigated the rise in serum T4 induced by a single dose of bovine TSH in the three sets of recombinant inbred lines that we previously investigated for induced Graves' hyperthyroidism. In addition, we tested the parental strains and the F1 hybrids of these families. Like many autoimmune conditions, Graves' disease develops predominantly in women. Consequently, our previous studies of induced Graves' disease, as well as our current investigations, were performed in female mice.
Materials and Methods
Mouse strains
Female mice of the following strains (5–9 weeks old) were obtained from the Jackson Laboratory:
1. Parental strains C3H/HeJ, A/J, BALB/cJ, and C57BL/6J; the latter two are the most closely related available to the parental strains of the CXB set (BALB/cByJ and C5BL/6ByJ). Parental strains are abbreviated C3H, A, BALB, and B6.
2. F1 hybrids: B6C3F1/J (B6 female X C3H male), CB6F1/J (BALB female X B6 male), and B6AF1/J (B6 female X A/J male). T4 levels of parental strains and F1 progeny are of great interest, but are not used in computing genetic linkage.
3. AXB/PgnJ 1, 2, 4, 5, 6, 8, 10, 12, 13, 15, 19a, 23, 24; BXA/PgnJ 1, 2, 4, 7, 8, 11, 12, 13, 14, 16, 24, 25, 26 (abbreviated A1 to A24 for AXB strains and B1 to B26 for BXA strains; as a group, they are referred to as “AXBXA”).
4. CXB/ByJ 1 to 7, CXB/HiAJ 8 to 13 (abbreviated CXB1 to 13).
5. BXH TyJ 2, 4, 6–11, 14, 19, BXH KccJ 20 and 22 (abbreviated BXH2, 4 etc. to BXH22).
Mice from the Jackson Laboratory were fed LabDiet 5K52/5K67 or LabDiet 5K54 (containing 2.1 and 2.2 ppm iodine, respectively). All mice were maintained at the Cedars-Sinai Medical Center until 7–9 weeks old (on average); during this time (1–3 weeks), they were fed LabDiet Mouse Chow 5015 (1.01 ppm iodine) ad libitum. The mean (±standard deviation [SD]) ages of these recombinant inbred families was 7.7±1.0 weeks for CXB; 7.9±0.6 for BXH; 8.9±1.0 for AXB and BXA. Strains exceeding the 90th percentile for age in each set were CXB2 (9.9 weeks), BXH11 (9.1 weeks), AXB4 and AXB23 (11.3 and 11.1 weeks, respectively). All use of mice was approved by the Institutional Animal Care and Use Committee and performed with the highest standards of care in a pathogen-free facility.
L-T3–induced suppression of T4 and TSH stimulation
T4 release induced by TSH was studied in L-T3 suppressed mice based on the protocol of Maia et al. (12) as previously described (11). L-T3 for i.p. injections was prepared as follows: 2 mg L-T3 (T6397; Sigma-Aldrich) was dissolved in 1 mL 0.1 N NaOH, diluted 1: 40 (0.2% bovine serum albumin in phosphate-buffered saline) and the pH adjusted to 7.0; 0.1 mL of this solution (containing 5 μg L-T3) was injected.
The schedule for injections and blood drawing (Fig. 1) was as follows: Day 0, mice were weighed and bled from the tail vein to provide baseline T4 values; Days 1, 2, and 3, each morning, L-T3 (5 μg/mouse) was injected i.p.; Day 3, afternoon, blood was drawn from the tail vein to establish the degree of L-T3–induced suppression on T4; Day 4, bovine TSH (10 mU; Sigma-Aldrich) was injected i.p. and mice were euthanized 5 hours later. Variability in the response of mice to i.p. injections has been documented (13,14). To reduce such variability to a minimum, all mice were handled and injected by the same experienced investigator (S.H.).

Schedule for L-triiodothyronine (L-T3) administration i.p. to suppress baseline thyroxine (T4) before injecting bovine thyrotropin (TSH; 10 mU) to induce T4 secretion in mice. The measurement parameters used for genetic linkage analysis in recombinant inbred mice are included.
Serum T4
Total T4 was measured by radioimmunoassay in sera (25 μL) from individual mice (Coat-a-Count total T4 kit; Siemens Health Diagnostics, Deerfield, IL). Standards ranged from 1–24 μg/dL T4 (sensitivity 0.25 μg/dL). For each mouse strain, sera for all time intervals (baseline, after L-T3 treatment and at postmortem after TSH injection) were tested in the same assay. T4 levels were computed from kit standards and reported as μg/dL. The following T4 measurements were examined for genetic linkage (summarized in Fig. 1): (i) Baseline T4; (ii) T4 post–L-T3 suppression; (iii) T4 release (T4 after TSH challenge minus T4 post–L-T3 suppression); (iv) percentage suppression by L-T3 calculated from (baseline T4 − T4 post–L-T3 suppression)/baseline T4×100%; and (v) T4 release as % baseline T4 (calculated from T4 release/baseline T4×100%).
Baseline TSH
Baseline TSH levels were determined by Dr. Roy Weiss (Thyroid Unit, University of Chicago, IL; fee for service) by radioimmunoassay (15). For this purpose, pooled samples from 4–5 mice were analyzed in duplicate aliquots of undiluted serum (50 μL). Sera with values <10 mU/L were assigned an arbitrary value of 5.0 for linkage analysis.
Statistical analysis and linkage analysis
Comparisons between traits in different mouse strains were performed by analysis of variance (ANOVA; SigmaStat, Jandel Scientific Software). In calculating the mean and standard errors, the value 0.001 was assigned to mice with undetectable T4 levels after L-T3 and for mice in which there was no rise in serum T4 after TSH.
Quantitative trait loci (QTL) for the traits summarized in Figure 1 were mapped using genotype files for AXB/BXA, BXD, BXH, and CXB strains generated by Williams et al. (16) and embedded in GeneNetwork (GN) (17). The probability of linkage between T-T4 and F-T4 and previously mapped genotypes was estimated at ∼1 centiMorgan intervals (∼2 megabase [Mb]) along the entire genome, except for the Y chromosome. To establish criteria for suggestive and significant linkage, a permutation test was performed (24,000 permutations at 1-centiMorgan intervals) (18). This test compares the peak likelihood ratio statistics (LRS; LRS=LOD×4.6, where LOD is the logarithm of the odds) obtained for a given data set with the peak LRS score obtained for the same data set.
The phenotypes have been entered into the GN databases (18) under the following trait accession identifiers: AXB/BXA GN 10207 through 10213, 10256; CXB GN 10692 through 10698, 10742; BXH GN 10162 through 10169. These data can be found by searching the CXB, BXH or AXB/BXA databases for the name “McLachlan.” The GN trait numbers are provided to enable readers to verify and extend the data analysis. In addition, GN interval maps connect directly to ENSEMBL and to the University of California Santa Cruz Genome Browsers and it is comparatively easy to study the gene complement of the QTLs.
Results
Suppression by L-T3 and TSH-induced T4 release in parental strains and F1 offspring (all females)
We investigated serum T4 suppression by three daily i.p. injections of L-T3 (5 μg) and the release of T4 following one i.p. injection of bovine TSH (10 mU) in the parental strains and F1 offspring (all females) of three recombinant inbred sets as follows: BXH parental strains and F1 offspring (termed B6, C3H, and B6C3F1); CXB parental strains and F1 offspring (BALB, B6, and CB6F1); AXBXA parental strains and F1 offspring (A, B6, and B6AF1). As mentioned, we tested the Jackson (J) strains of BALB/c and B6, the most closely related strains to the parental CXB Bailey (By) strains. The latter would require resuscitation and breeding from cryopreserved embryos.
Baseline T4 values were significantly lower in B6 mice than in C3H, BALB, or their F1 offspring. However, A and B6AF1 offspring had lower levels similar to B6 mice (Fig. 2A). L-T3 administration suppressed serum T4 by 80% in C3H mice and by greater than 85% in the other strains (Fig. 2B). T4 release induced by TSH was lowest in A and B6 mice compared to more robust responses in BALB/c and all three sets of F1 offspring, including those derived from parental A and B6 strains (Fig. 2C).
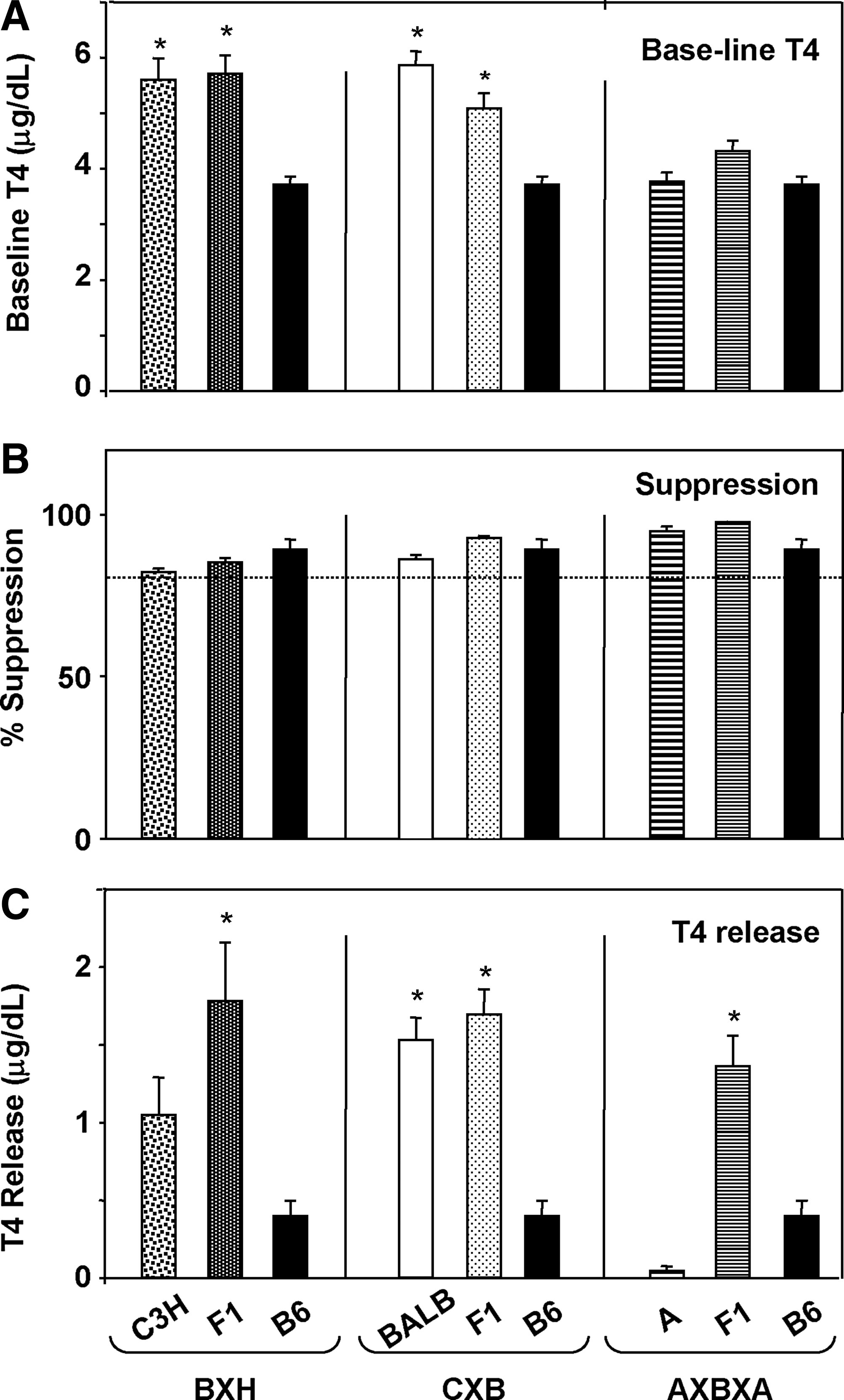
Baseline T4, L-T3–induced suppression and TSH induced release of T4 in the parental strains and F1 cross for three recombinant inbred sets: BXH (parents B6 and C3H; cross B6C3F1); CXB (parents BALB and B6; cross CB6F1); and AXBXA (parents A and B6, cross B6AF1). The data for B6 mice are repeated in each panel to provide a direct comparison with the other parents (C3H, BALB, and A). Bars are the mean+SEM for 3–5 mice in each strain.
It should be emphasized that all our studies, including parental and F1 strains and recombinant inbred strains, were performed in female mice. Given the potential effects of the estrous cycle and sex differences in some parameters of the thyroid axis, our results may not be applicable to males.
Suppression by L-T3 and responses to TSH in three recombinant inbred sets
Previously, we observed that orally administered L-T3 only suppressed T4 levels in large outbred mice and inbred males, whereas L-T3 injected i.p. was effective for inbred female mice (11). Consequently, in the present study, L-T3 suppression was performed by three daily i.p. injections of L-T3 (5 μg). Our protocol was based on that of Maia et al. (12) showing that, in approximately 7-week-old male mice, serum T4 levels were reduced 24 hours after the last of three L-T3 (5 μg) injections. Moreover, to ensure that the mouse weight was not a genetic susceptibility factor influencing L-T3 suppression and/or the subsequent response to TSH, mice were weighed at the start of the experiments.
Despite variability in body weight, L-T3 effectively suppressed T4 levels by>85% in all CXB and BXH strains (Fig. 3A) and AXB/AXA strains (hereafter called “AXBXA”) with one exception (BXA16, broken arrow, Fig. 3B). However, several strains in each of the three recombinant inbred sets did not release T4 in response to TSH (Fig. 3A, B).

Weight, T4 at baseline and post–L-T3 administration, % suppression, and T4 release in response to TSH in three sets of recombinant inbred mice: CXB (13 strains) and BXH
Genetic linkage for baseline parameters: body weight, T4, and TSH
QTL were mapped using the CXB, BXH, and AXBXA database files (16,19) for the following parameters: body weight, serum T4, and TSH (all at baseline), T4 post–L-T3 suppression, % L-T3 suppression of serum T4, and three measurements of the response to bovine TSH in L-T3–suppressed mice (Fig. 1). The outcome of linkage analysis for baseline parameters is described next and the genetic susceptibility for L-T3 suppression and TSH-induced T4 release is reported in the subsequent section.
Body weight was suggestively linked to loci on chromosomes (Chr) 12, 17, and 10 in the BXH, AXBXA, and CXB sets, respectively (Table 1). Mice of these strains were previously studied for body weight, generally in much younger mice than those used in our investigation. Also, some earlier investigations involved only six or seven CXB strains and, therefore, linkage analysis could only be performed by including data for the parental strains (Supplementary Table S1; Supplementary Data are available online at
For each trait studied, the following information is provided: recombinant inbred set (number of strains), likelihood ratio statistic (LRS) suggestive of linkage (significant linkages in
Cd180 (103.48364 Mb), CD180 antigen; Dio3 (111.517 Mb), deiodinase type 3; EG621312 (103.94987 Mb), predicted gene, EG621312; Epb4.113 (69.50615 Mb), erythrocyte protein band 4; Grem1 (33.01 Mb), gremlin 1 (human 15q13.3); Pik3r1 (102.450717 Mb), phosphatidylinosital 3 kinase regulatory subunit 1; Lama1 (68.0466 Mb), laminin alpha 1; Lrrfip2 (111.02064 Mb), leucine-rich repeat (in FLII) interacting protein 2; Pip5k1b (24.36929 Mb), phosphatidylinositol-4-phosphate 5-kinase, type 1 beta; Rab18 (6.7652 Mb), member of Ras oncogene family.
Baseline T4 was linked to Chr 2 in the AXBXA set and to loci on Chr 9 and Chr 19 in the CXB set (Table 2). The Chr 2 linkage for basal T4 that we previously observed in AXBXA strains (21) was located slightly downstream of the present locus (∼108 Mb versus ∼112 Mb; Supplementary Table S2). The major loci for baseline T4 in CXB strains in the present study differed from those reported earlier, but linkage was suggestive (LRS 7.40) for the locus on Chr 13 (∼104 Mb) highlighted previously (21). In the 12 BXH strains available (the 13th strain has been cryopreserved), we found no linkage for baseline T4 levels.
For each trait studied, the following information is provided: recombinant inbred set (number of strains), LRS value suggestive of linkage (significant linkages in
T4 release=T4 after TSH−T4 after L-T3.
Without BX9 (hypothyroid; see text).
Excluding BXA16 (incomplete L-T3 suppression).
Cd180 (103.483634 Mb), CD180 antigen; Gnas (174.1089821 Mb), guanine nucleotide binding protein, alpha stimulating complex locus; Lasc1, uncharacterized protein 4931422A14; Pik3r1 (102.450717 Mb), phosphatidylinositol 3 kinase; Pou4f1 (104.86154 Mb), Pou domain class 4 transcription factor 1; Pouf3f2 (22.4139 Mb), Pou domain class 3 transcription factor 2; Rgs19 (181.423127 Mb), regulator of G-protein signaling 19; Trhr2 (124.8807 Mb), thyrotropin releasing hormone receptor 2; Trim52 (106.50541 Mb), tripartite motif-containing 52; Tshz2 (169.45915 Mb), t-shirt zinc finger family.
Serum TSH was undetectable in serum pools from most recombinant inbred strains, all females. These findings are in agreement with previous studies for females of other strains [for example, Ref. (15)]. Only 3 of 25 AXBXA and 2 of 13 CXB strains had TSH levels greater than 10 mU/L (the assay detection limit). Therefore, linkage analysis could not be performed for these two recombinant inbred sets. However, linkage analysis was performed for the BXH set in which 6 of 12 strains had detectable TSH levels. TSH in female BXH mice is tentatively linked to a Chr 18 locus close to Rab18, a member of the Ras oncogene family (Table 1).
Analysis of serum TSH provided other unexpectedly valuable information. In the BXH9 strain, TSH was much higher than in the other 5 BXH strains with detectable TSH levels, and far exceeded TSH levels in the CXB and AXBXA strains in which TSH was also detectable; namely, 80 mU/L in BXH9 versus 38.4 mU/L (mean+2SD) in the other 10 strains. The BXH9 strain also had the lowest serum T4 for this set (3 μg/dL, Fig. 3A), reflecting subclinical hypothyroidism. Mice of this strain were unresponsive to endogenous TSH and, as we observed, to bovine TSH. Therefore, linkage analyses of the T4 response to TSH in BXH strains were performed by excluding BXH9 strain data.
Genetic basis for L-T3–induced suppression of T4- and TSH-induced release of T4
In CXB strains, the serum levels of T4 post–L-T3 suppression were linked (although with low LRS values) to loci on Chr 14 and 8 (Table 2) and the percentage suppression of T4 by L-T3 was linked to similar Chr 8 loci (Supplementary Table S3). No linkage was observed for these traits in the AXBXA and BXH sets.
Because our specific interest was in determining the genetic basis for the T4 response to a single injection of bovine TSH in L-T3–suppressed mice, we focused on the linkage analysis of the following traits: (i) T4 levels attained after TSH; (ii) T4 release (T4 after TSH minus T4 post–L-T3 suppression); and (iii) T4 release expressed as % baseline T4. The major findings are as follows:
1. CXB set (13 strains): T4 release was strongly linked (LRS 20.683) to loci on the distal ends of Chr 2 and 13 (Table 2 and Fig. 4). T4 levels after TSH were linked to the same loci as for T4 release, but with lower LRS values (Supplementary Table S3). T4 release expressed as % baseline continued to be linked to Chr 2 (but not Chr 13) and the loci involved were different (Supplementary Table S3).
2. BXH set (11 strains, excluding BXH9): T4 release and T4 after TSH were linked to loci on Chr 2 and 7 and (with a lower LRS value) to loci on the distal terminus of Chr 4 (Table 2 and Supplementary Table S3). The trait T4 release expressed as % baseline was linked to a Chr 1 locus, as well as maintaining linkage (with lower LRS values) to the same Chr 2 and Chr 7 loci (Supplementary Table S2).
3. AXBXA set (25 strains; excluding the strain BXA16, which was not effectively suppressed by L-T3): All three traits, namely, the T4 release expressed as % baseline T4, and T4 after TSH, were linked to similar loci on the proximal end of Chr 4 (Table 2 and Supplementary Table S3).
For all three recombinant inbred sets, the trait giving the highest LRS values was the T4 release. To eliminate the possible effect of increased age, the linkage analyses were repeated for this trait for each recombinant inbred set excluding the strains with ages exceeding the 90th percentile for each set; namely, CXB2 (9.9 weeks), BXH11 (9.l1 weeks) and both AXB4 and AXB23 (11.3 and 11.1 weeks, respectively). After excluding these strains, the LRS values were reduced from 20.333 to 18.135 (CXB), 16.470 to 15.360 (BXH), and 14.284 to 11.848 (AXBXA). Importantly, however, linkage was maintained and the Chr and loci identified were unchanged (Table 2, italics).
To summarize, the genetic susceptibility to T4 release was linked to Chr 2 loci in CXB and BXH strains and Chr 4 loci in AXBXA and BXH strains. The Chr 2 loci linkage for T4 release in CXB and BXH strains, although to the same distal terminus, were discrete and nonoverlapping (Fig. 4), separated by 3.437 Mb. As already indicated, the Chr 4 linkage was to loci on the proximal end in the AXBXA set and to the distal terminus in the BXH set. Overall, distinct linkage patterns were observed for these traits in the three recombinant inbred sets with no chromosomal overlap.
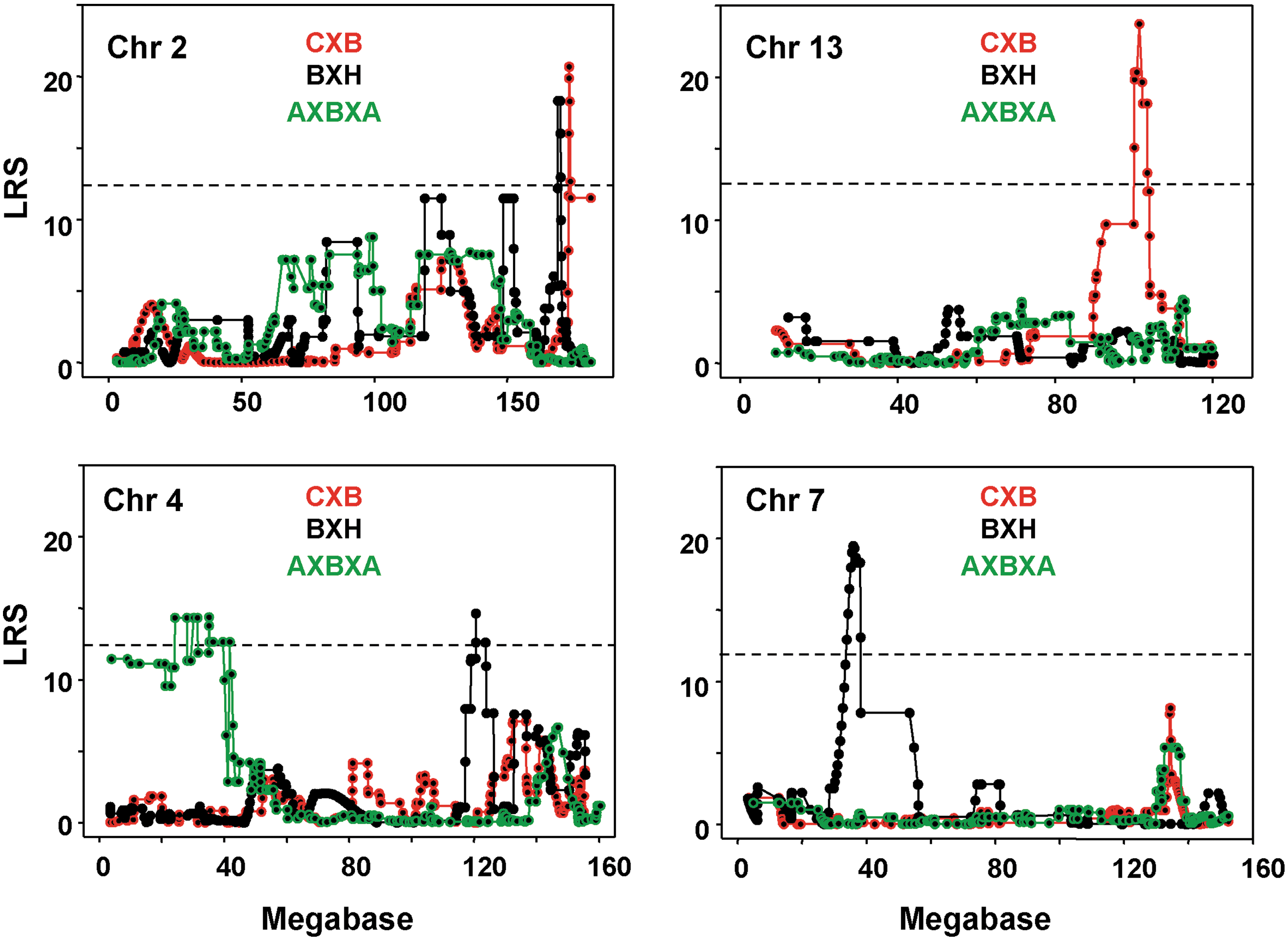
Interval mapping on chromosomes 2, 13, 4, and 7 for T4 released in response to bovine TSH in CXB, BXH, and AXBXA mice. Likelihood ratio statistic (LRS) values are on the vertical axis; Mb on the horizontal axis. The broken horizontal lines indicate the mean LRS value above which there is a suggestive linkage for the three recombinant inbred sets (mean 12.2 from CXB 13.3, BXH 12.6, and AXBXA 10.7). GeneNetwork (GN) identifications for CXB (GN10694), BXH (GN 10166); AXBXA (GN10211). Number of strains studied: CXB, 13; BXH, 11; AXBXA, 25.
Genetic susceptibility for TSH-induced T4 release compared with genetic linkage for elevated T4 induced by A-subunit adenovirus immunization
Previously, we investigated the genetic basis for hyperthyroidism in recombinant inbred mice immunized with TSHR A-subunit adenovirus to induce hyperthyroidism (3,5,6). There were no similarities, or overlap, between the loci linked to induced hyperthyroidism (Table 3) and those we observed in the present study to be linked to the response to TSH in L-T3–suppressed mice of the same three recombinant inbred sets (Table 2 and Supplementary Table S3).
Traits examined include serum T4 after two or three immunizations (2×or 3×) as well as the difference between T4 after immunization and baseline T4 (“delta T4”). Suggestive LRS values are provided, together with the LOD values (LOD=LRS/4.61), the relevant Chr, location in Mb, and the corresponding [or likely] human Chr. References: (3,5,6).
Discussion
We investigated the genetic basis for the magnitude of the T4 response to a single dose of bovine TSH in three sets of L-T3–suppressed recombinant inbred mice, CXB, BXH, and AXBXA. All three sets share B6 as one parental strain. Consistent with some previous findings, baseline serum T4 values (22,23) and the response to TSH stimulation (11) were lower in parental B6 and A than in BALB and C3H mouse strains (22,23). Unexpectedly, T4 release was high in all three sets of F1, including the F1 of the low responder A and B6 parents, suggesting hybrid vigor.
An important issue is why we studied female rather than male mice. First, Graves' disease predominantly affects women rather than men. Consequently, our studies of the genetic basis for induced murine hyperthyroidism were performed in female mice (3,5,6). Second, it is advantageous to investigate mice of the same sex when comparing the genetic basis for the T4 response to TSH relative to our previous studies on induced autoantibody-mediated hyperthyroidism. Indeed, a recent study of severe murine influenza identified sex-specific candidate genes in recombinant inbred mice (23), confirming the importance of using mice of the same sex for genetic studies.
In recombinant inbred lines, we measured the mouse weight, serum T4, and TSH at baseline. In our earlier study, body weight played a critical role in the efficacy of T4 suppression using L-T3–supplemented drinking water (11), rather than L-T3 injected intraperitoneally as in the present study. In other investigations, body weight had been investigated, as one parameter, in the CXB, BXH, or AXBXA sets (20,24 –28). In AXBXA strains (but not CXB or BXH sets), body weight was linked to the same Chr 17 locus identified in a previous study (20). It is important to emphasize that, although some marker genes are provided, no candidate genes for body weight were suggested by the present or earlier investigations.
The major CXB loci for baseline T4 in the current study differed from those we reported earlier, which were located on Chr 1, 2, 11, and 13 (21) although linkage was suggestive for a previously identified Chr 13 locus (21). No linkage for baseline T4 was observed for the 12 BXH strains in the present study. In the AXBXA set, baseline T4 was linked to a Chr 2 locus slightly upstream of the locus highlighted previously (21). Few previous studies examined the genetic basis for T4 levels alone rather than in relation to another trait, such as audiogenic seizures (29) or as contributing to the life span (for example (30). In UM-HET3 mice, derived by crossing the F1 of (BALBc×B6) with the F1 of (C3H/He×DBA/2), loci that affected T4 levels were located on Chr 4, 15, and 17 (30). None of these Chr or loci were similar to those we observed for baseline T4 summarized on page 7 or for the response to injected TSH (to be discussed below).
Our observations for serum TSH are limited because, as explained above (Introduction) we studied female mice in which TSH is usually undetectable [for example, Ref. (15)]. Previous studies in male rats demonstrated linkage between TSH and two chromosomal intervals; one included the genes for the TSH receptor and type 2 deiodinase and the other included the type 1 deiodinase gene (32). In a subsequent study using consomic rats, the higher TSH levels were associated with reduced expression of the TSHR, reflecting an adjustment of the feedback loop to reduced TSH sensitivity (33). In female BXH mice, the only recombinant inbred set for which analysis was possible, serum TSH was tentatively linked to a Chr 18 locus with no obvious candidate genes.
Suppression of serum T4 by i.p. injection of L-T3 was effective in all except one recombinant inbred strain (BXA16). Not surprisingly, therefore, no linkage was observed for serum T4 after L-T3 administration, or for the percentage of T4 suppression, in AXBXA or BXH strains. However, in the CXB set, the same traits were linked, with low LRS values, to loci on Chr 14 and 8. T4 suppression by L-T3 is likely associated with the hypothalamus/pituitary function. Accordingly, in the CXB set, T4 suppression by L-T3 was suggestively linked, although with a low LRS value to a Chr 8 locus near the gene for the TSH releasing hormone receptor 2 (Trh2).
Despite effective L-T3 suppression of serum T4 levels by >85% in virtually all CXB, BXH, AXB/AXA mice, several strains in each of the three recombinant inbred sets did not release T4 in response to bovine TSH. Of interest, in animals not subject to L-T3 suppression of endogenous TSH, recombinant human TSH readily induced T4 release from CRL:CF-1BR male mice, but not in male Sprague-Dawley rats (34).
We analyzed three traits for the response (in L-T3–suppressed mice) to TSH, namely: serum T4 after TSH, T4 release (the difference between T4 after TSH and T4 post–L-T3 suppression), and T4 release expressed as a percentage of basal T4 levels. Of these three traits, the highest LRS values were obtained for T4 release. Moreover, the strongest linkage for T4 release was in the CXB and BXH sets (13 and 12 strains studied), not in the larger AXBXA set (25 strains studied). This finding reflects the divergence of responses to TSH in the parental strains, BALB or C3H versus B6 compared with similar responses in parental B6 and A strains. The observation also suggests that the divergent parental strain responses may sometimes have more impact than the statistical power provided by the increased size of a recombinant inbred set.
In CXB strains, T4 release was strongly linked (LRS 20.683) to loci on the distal ends of Chr 2 and 13. In the BXH set, the same trait was linked to loci on Chr 2 and 7 and (with a lower LRS value) to loci on the distal terminus of Chr 4. Although T4 release was linked to the distal end of Chr 2 in both CXB and BXH sets, the loci were discrete with no overlap. In AXBXA strains, T4 release was linked to the proximal region of Chr 4, different from the BXH Chr 4 loci at the proximal end. As already emphasized, given the potential effects of the estrous cycle and sex differences in some parameters of the thyroid axis, our results may not be applicable to males. Overall, as we previously observed for baseline T4 levels (21), the linkages for T4 release were distinct in the three sets of recombinant inbred mice. Although T4 release is conceptually simple, several parameters are involved, including baseline T4 levels and the ability to respond to TSH. The linkage variability in different sets of recombinant inbred mice parallels the findings for ethnic variations in humans.
There was no relationship between strains showing the greatest release of T4 induced by TSH (present study) and the increase in T4 after two or three immunizations with A-subunit adenovirus to induced Graves' disease (3,5,6). Some discrepancies are readily explained. For example: TSH-induced T4 release was similar in BXA12 versus BXA13 (Fig. 3B, indicated by brackets). In contrast, after immunization, BXA12 mice were euthyroid, whereas BXA13 mice developed extreme hyperthyroidism (3). This difference arises because, unlike BXA13 strains, BXA12 mice were unable to generate TSHR antibodies in response to immunization. In addition, it should be emphasized that the present study involves the acute release of T4 in response to TSH. In contrast, the immunization protocol takes place over a period of 6 weeks and hyperthyroidism develops one to 2.5 months after the initial immunization.
A major issue is whether there is any relationship between the loci or genes we observed and those linked to the thyroid function in humans. The T4 response to TSH is a parameter associated with the thyroid gland function and the genes highlighted by our investigation are in line with those controlling the thyroid function in humans. A number of genetic loci play a role in the levels of TSH, thyroid hormones and thyroid volume and goiter in humans (35 –40) (summarized in Table 4). To our knowledge, none of the murine genes we observed were the same as in humans. However, two linkages in mice are of interest. It must be emphasized that confirmation of a role for any of these putative genes/loci will require further investigation. First, one of the two strongest linkages for T4 released after TSH in CXB mice is to regions on Chr 13 between the genes PDE8B and PDE4D (encoding phosphodiesterase 8B and 4D, respectively; Table 4). Accumulating evidence points to PDE8B as a gene responsible for TSH levels in humans (35,39). Because the LRS peaks from our study do not correspond to the locations of PDE8B and PDE4D, it is possible that other, as yet, unidentified genes in this region may play a role. Second, the other strong linkage for T4 released after TSH in CXB mice is to a Chr 2 region close to the GNAS1 gene (guanine nucleotide-binding protein, alpha-subunit), also shown to play a role in controlling TSH levels in humans (35).
Included in italics are the gene locations for the strongest linkage data for T4 levels or release in response to thyrotropin in CXB strains (mouse Chr 2 and 13; LRS values 20.331).
It should be appreciated that in humans, multiple loci may contribute to the same trait, as illustrated for TSH in Table 4. Although some loci have been replicated in additional cohorts [for example, Refs. (35,40)], the same trait may be controlled by different loci in different ethnic groups: for example, the variable role of the THRB gene and TSH levels [reviewed in Ref. (30)] and the well-known ethnic differences in human leucocyte antigen (HLA) associations in autoimmune thyroid disease [reviewed by Jacobson et al. (41)]. These ethnic variations in human genetic susceptibilities parallel the different genetic linkages in recombinant inbred mice, derived from B6 parents bred to C3H/He, BALB/c, or A strains, which we previously observed for baseline T4 levels (21), induced hyperthyroidism (3,5,6), and, as we report here, for TSH-induced T4 responses.
In conclusion, T4 release in response to TSH was more strongly linked in CXB and BXH sets, derived from parental strains with widely divergent T4 release values, than in AXBXA strains, which were generated from parents with similar TSH-induced responses. The genetic susceptibility for thyroid sensitivity, measured as T4 released in response to bovine TSH, was distinct for three sets of recombinant inbred mice, at least in females, despite the fact that they share the C57BL/6 parental strain. There was no relationship between Chr or loci linked to induced hyperthyroidism (which develops over months) versus those linked to the acute TSH-induced T4 response. In mice, as in humans, multiple loci contribute to the same trait and different loci are observed in different ethnic groups for the same trait (38). In addition, evidence supports the concept that “human disease genes can be identified through cross-species gene mapping of evolutionary conserved processes” (42). Consequently, our findings for loci that overlap with, or are located upstream or downstream of putative human TSH genes, suggest that novel loci responsible for controlling thyroid function in humans may yet be revealed.
Footnotes
Acknowledgments
This work was supported by the National Institutes of Health Grants DK82390 and DK54684 (S.M.M.), DK19289 (B.R), U01AA13499, U24AA13513, P20-DA-21131, U01CA105417, and U24RR021760 (R.W.W.). We are also grateful for support by Boris Catz.
Disclosure Statement
No competing financial interests exist.