
Abstract
Background:
Anaplastic thyroid cancers (ATCs) represent only 1%–2% of all thyroid tumors, but they account for up to 50% of the mortality. Treatment of differentiated thyroid carcinomas is well standardized and the use of radioiodine represents an essential step; in contrast, there is no standardized therapeutic approach for anaplastic tumors and their prognosis is poor. The resistance of ATC to radioiodine treatment is principally due to the absence of expression of the sodium iodide symporter (NIS), mainly due to epigenetic silencing. The acetylation status of histones is involved in the epigenetic control of gene expression and is usually disrupted in advanced thyroid cancer. Histone deacetylase inhibitors have been demonstrated as potent anticancer drugs with several different effects on cell viability and differentiation.
Methods:
Stabilized ATC cell lines (BHT-101 and CAL-62) and primary cultures from patients who underwent thyroidectomy for ATC were treated with the histone deacetylase inhibitor LBH589. After treatment, we evaluated the expression and function of NIS. Gene expression was evaluated by real-time polymerase chain reaction (RT-PCR), NIS promoter activity was determined with a luciferase reporter assay, and protein expression was assessed through immunofluorescence. We tested the protein function by 125I uptake and efflux experiments; finally the cytotoxic effect of 131I was determined with a clonogenic assay.
Results:
Our results demonstrate that treatment with LBH589 leads to NIS RNA expression as shown by RT-PCR and luciferase assay, and to protein expression as determined by immunofluorescence in vitro and by immunohistochemistry in xenograft tumors. Moreover, 125I uptake and efflux experiments show the correct protein function and iodine retention, which translate into cytotoxicity effects, as demonstrated by a clonogenic assay with 131I.
Conclusions:
This study supplies a new potential strategy for the treatment of ATC by modifying gene expression with the aim of inducing responsiveness towards radioiodine therapy.
Introduction
In the “epigenetic progenitor model” (3), epigenetic alterations are considered to be equally if not more important than mutational events for cancerogenesis, leading to the concept of epigenetic therapy. Indeed, there is a strong basis for the use of “epigenetic modifier drugs” to modulate the epigenome of cancer cells to a status resembling the one seen in normal cells, thereby leading to tumor cell death or restoration of normal differentiation (4).
The presence of the sodium iodide symporter (NIS) is both a diagnostic and a therapeutic tool in the management of thyroid cancer due to its ability to mediate iodide uptake (5). Unfortunately, approximately 10%–20% of thyroid tumors comprising metastatic differentiated thyroid carcinomas, as well as poorly differentiated and anaplastic cancers (6), fail to take up radioiodine and are unable to concentrate enough 131I for effective cytotoxic therapy; tumors with reduced or absent NIS activity are therefore generally associated with a poor prognosis.
Deacetylase inhibitors (DCIs) are compounds that can alter the chromatin acetylation status, modulating the transcription of about 2% of genes involved in growth, differentiation, and apoptosis (7). NIS gene silencing has been related to histone acetylation. The use of DCIs like trichostatin A (TSA), sodium butyrate (NaB), and valproic acid restored NIS expression in human thyroid cancer cell lines (8 –10). Moreover, increased NIS expression was demonstrated in nonthyroid cell lines after treatment with TSA, NaB (10), and LBH589 (11). As far as ATC is concerned, we have reported elsewhere that valproic acid increased NIS expression in poorly differentiated thyroid cancer cell lines (9), but the effect of the drug in ATC is limited to mRNA expression without any impact on iodine uptake. Panobinostat or LBH589 is a potent pan-DCI with anticancer activity as demonstrated in vitro against classical Hodgkin's lymphoma (cHl), and in clinical phase I and phase II trials in patients with relapsed cHl (12). We recently demonstrated that LBH589 affects histone acetylation and has potent cytotoxic and anti-invasive activities in in vitro and in vivo ATC (13,14).
The aim of the present work was the evaluation of the ability of LBH589 to induce NIS expression and activity in ATC in order to obtain significant radioiodine uptake and sensitivity.
Materials and Methods
Cell lines and reagents
ATC (BHT-101 and CAL-62) cell lines were purchased from Deutsche Sammlung von Mikroorganismen und Zellkulturen (Braunschweig, Germany). Both cell lines were maintained at 37°C, in 5% carbon dioxide and 95% humidity, in Dulbecco's modified Eagle's medium (D-MEM)-F12 (Invitrogen, Groningen, The Netherlands), with 100 IU/mL penicillin and 100 μg/mL streptomycin added, supplemented with 10% heat-inactivated fetal calf serum (FCS; Euroclone, Wetherby, United Kingdom). LBH589 was provided by Novartis Pharma AG (Basel, Switzerland), prepared as a 5 mM stock solution in dimethylsulfoxide (DMSO) and stored at −80°C.
Primary culture establishment
ATC samples were obtained from seven patients undergoing thyroidectomy for ATC (cultures were labeled CPa–CPg). Samples obtained from two healthy thyroids were used as controls (labeled CPS1 and CPS2). Informed consent for the study was obtained from each patient. Tissues were obtained under sterile conditions and processed within 24 hours of surgery. Samples were washed three times in saline containing penicillin/streptomycin and nystatin and then minced in D-MEM-F12 and incubated overnight with 0.25% trypsin at 4°C. After this, enzymatic digestion was performed using 0.35% collagenase (Sigma, Milan, Italy) at 37°C for 60 min. Cells were filtered through double layers of sterile gauze and washed twice with D-MEM-F12 plus 20% FCS. Cells were cultured in complete medium containing 100 IU/mL penicillin and 100 μg/mL streptomycin, 50 μg/mL gentamicin, 2.5 μg/mL amphotericin B, and 20% FCS.
Primary cultures were tested for cytokeratins by immunofluorescence (monoclonal Anti-pan Cytokeratin Clone C-11; Sigma) and for thyroglobulin (TG), thyroid transcription factor 1 (TTF-1) and NIS expression by real-time polymerase chain reaction (RT-PCR). All the cultures showed complete concordance with the original tumors or normal thyroid tissues (Table 1).
TTF1, thyroid transcription factor 1; NIS, sodium iodide symporter; Neg, negative; Pos, positive; M, male; F, female; ATC, anaplastic thyroid carcinoma.
Gene expression evaluation with RT-PCR
Cells (1×106) were seeded in 75 cm2 flasks and treated with increasing doses of LBH589 (0–50 nM). After 24 hours of treatment, total RNA was extracted using TRIzol Reagent (Invitrogen Ltd., Paisley, United Kingdom). DNase I was added to remove remaining genomic DNA. One microgram of total RNA was reverse-transcribed with iScript cDNA Synthesis Kit (BioRad Laboratories, Inc., Segrate, Italy), following the manufacturer's protocol. Primers (Table 2) were designed using Beacon Designer 5.0 software according to parameters outlined in the BioRad iCycler Manual. The specificity of the primers was confirmed by BLAST analysis. RT-PCR was performed using a BioRad iQ iCycler Detection System with SYBR green fluorophore. Reactions were performed in a total volume of 25 μL containing 12.5 μL of IQ SYBR Green Supermix (BioRad Laboratories, Inc.), 1 μL of each primer at 10 nM concentration, and 5 μL of the previously reverse-transcribed cDNA template. The protocol for each primer set was optimized using seven serial 5×dilutions of template cDNA obtained from cells in basal conditions. The protocol used was as follows: denaturation (95°C for 5 minutes) and 40 cycles of amplification (95°C for 15 seconds, 60°C for 30 seconds). A melting curve analysis was performed following every run to ensure a single amplified product for every reaction. For each sample, all reactions were carried out at least in triplicate. Results were normalized using the geometric mean for three different housekeeping genes (β-actin, β2-microglobulin, and L13A) and expressed as relative expression fold versus the lower level of expression.
NIS promoter luciferase assay
Cells (2.5×105) were seeded in six-well plates. Twenty-four hours after seeding, cells were transfected with 1 μg of NIS-luc plasmid/well using Lipofectin Reagent (Invitrogen Ltd.). The plasmid, donated by Prof. G. Damante (Department of Biomedical Sciences and Technologies, University Hospital of Udine, Udine, Italy) contained 2.2 kb of 5′ genomic sequence of the NIS promoter linked to the luciferase (LUC) gene as reporter (15). After 24 hours, cells were treated with LBH589 (10–25 nM) for a further 24 hours. At completion luciferase activity was assayed with a Luciferase Assay System (Promega Corporation, Madison, WI).
NIS immunofluorescence microscopy
Cells (3×103) were seeded in 96-well plates. Twenty-four hours after seeding, cells were treated with LBH589 (25 and 50 nM) for 48 hours. After treatment cells were fixed in acetone/methanol (1:1) at −20°C for 20 minutes, washed with phosphate-buffered saline (PBS) containing 0.5% Triton X-100, 0.05% sodium azide (NaN3), and 1% bovine serum albumin and incubated overnight with a monoclonal anti-NIS antibody (1:1000; FP5A, Thermo Fisher Scientific, Fremont, CA) in PBS at 4°C. The cells were then washed three times with PBS containing 0.5% Triton and 0.05% NaN3 for 10 minutes followed by detection with an antimouse Cy3-conjugated secondary antibody (1:1000; GE Healthcare Europe, GmbH, Milan, Italy) in PBS plus 0.5% Triton and 0.05% NaN3 for 2 hours and the nuclei were counterstained with Hoechst 33258 (500 ng/mL in DMSO) in PBS. Cells were washed twice with distilled water and dried at 37°C.
125I uptake and retention by cells
Cells (2×104) were seeded in 24-well plates and incubated with LBH589 for 48 hours. 125I uptake was assayed as reported elsewhere (9). Briefly, after aspirating drug-containing medium, cells were washed with 1 mL of Hank's balanced salt solution (HBSS; Invitrogen). 125I uptake was initiated by adding 0.5 mL of HBSS containing 2 μCi carrier-free Na125I (PerkinElmer Life and Analytical Sciences, Inc., Boston, MA) and 30 μM sodium iodide (NaI). Samples were then incubated for 30 minutes at 37°C. At completion, the radioactive medium was removed, and the cells were then washed twice with ice-cold HBSS and solubilized with 1 mL of absolute ethanol for 20 minutes, after which cell-associated iodide was measured in a γ-counter (PerkinElmer). To avoid any unspecific iodide uptake, control cells were pre-incubated with 300 μM potassium perchlorate (KClO4) for 30 minutes and then treated with 125I, as described above.
Iodide efflux was studied by incubating cells for 30 minutes at 37°C in HBSS incubation buffer plus 2 μCi carrier-free Na125I (PerkinElmer) and 30 μM NaI. The medium was then replaced every 5 minutes with fresh HBSS, for 30 minutes. Afterwards, cells were solubilized with 1 mL of absolute ethanol. Radioactivity was counted in both the medium and the cells. Total radioactivity at the beginning of the efflux (100%) was calculated by adding the radioactivity of all the media to those of the cells. The percentage of activity remaining in the cells at different times was obtained by subtracting the activity at each time from that of the total radioactivity at the beginning (16).
Measured radioactivity was normalized by cell number and total activity in both the uptake and the efflux experiments.
131I cytotoxicity
Cells (1×102) were seeded into 24-well plates and left 48 hours to attach. Twenty-four hours after treatment with 25 nM LBH589, a clonogenic assay was performed. Briefly, drug-containing medium was discarded and cells were washed twice in PBS; the medium was then replaced with D-MEM-F12 (Invitrogen) in the presence or absence of 20 μCi 131I (GE Healthcare S.r.l., Milan, Italy) for 6 hours. At the end of the treatment the radioactive medium was discarded, and cells were washed and left in regular culture medium for the time corresponding to six doublings. Finally, cells that had formed colonies were fixed in methanol and stained with crystal violet. Control and 131I-treated colonies were counted (17).
Xenograft studies
Formalin-fixed, paraffin-embedded samples of tumors generated in SCID mice subcutaneously inoculated with CAL-62 cells and treated for 21 days with LBH589 (controls, n=5; 20 mg/kg LBH589, n=5), as described elsewhere (13), were assessed for the presence of NIS protein, using automated immunohistochemistry slide processing platforms (Ventana BenchMark AutoStainer [Ventana Medical Systems, Tucson, AZ] and Bond Max [Leica, Stuttgart, Germany]) and a primary monoclonal antibody against human NIS (Clone FP5A; Thermo Fisher).
NIS protein expression was evaluated by one of the authors (SA) using the following semiquantitative evaluation: 1) negative, no cells were stained; 2) positive, cells showed membrane, subnuclear, and/or cytoplasm stain.
Statistical analysis
Data are expressed throughout the text as means±SD, calculated from at least three different experiments. Comparison between groups was performed with analysis of variance (one-way ANOVA) and the threshold of significance was calculated with the Bonferroni test. Statistical significance was set at p<0.05.
Results
LBH589 effect on NIS expression
To investigate the effect of LBH589 on NIS expression, we performed RT-PCR on both stabilized and primary cell cultures. In BHT-101 and CAL-62 cells, LBH589 stimulated NIS expression in a dose-dependent manner (Fig. 1A and 1B); these results were further confirmed by the activation of a NIS promoter luciferase construct in both cell lines (Fig. 1C and 1D). LBH589 was also effective in inducing NIS expression in primary cell cultures, which did not express NIS in basal conditions (Table 1). In fact, we found NIS mRNA expression in all the cell lines treated with LBH589, albeit at different levels (Fig. 2A). As far as other thyroid-related genes (i.e., TTF-1, TG, TSH-R, and PAX8) are concerned, no modulation of the expression was observed (data not shown) after LBH589 treatment.
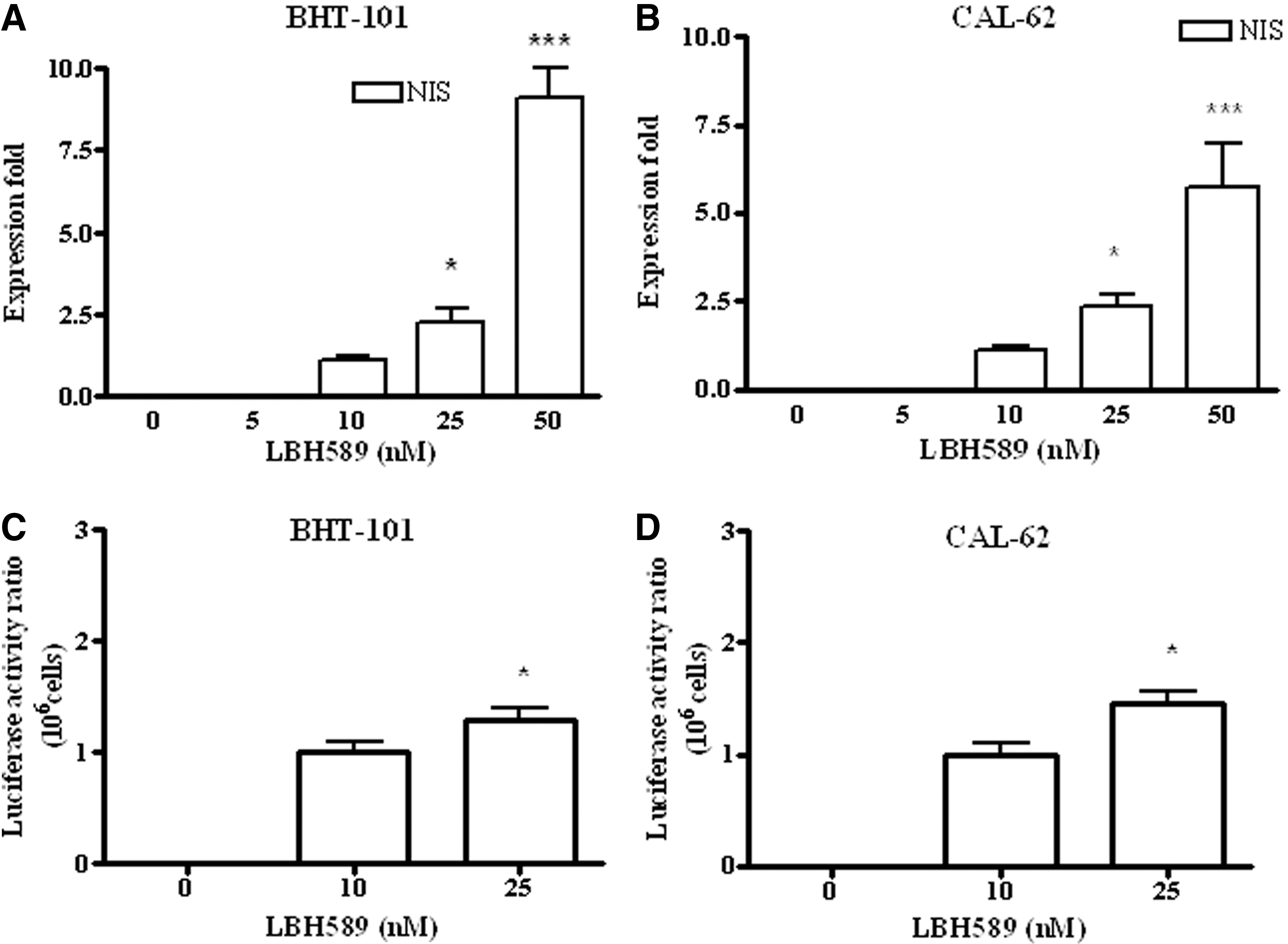
LBH589 effect on the expression of the NIS gene in BHT-101
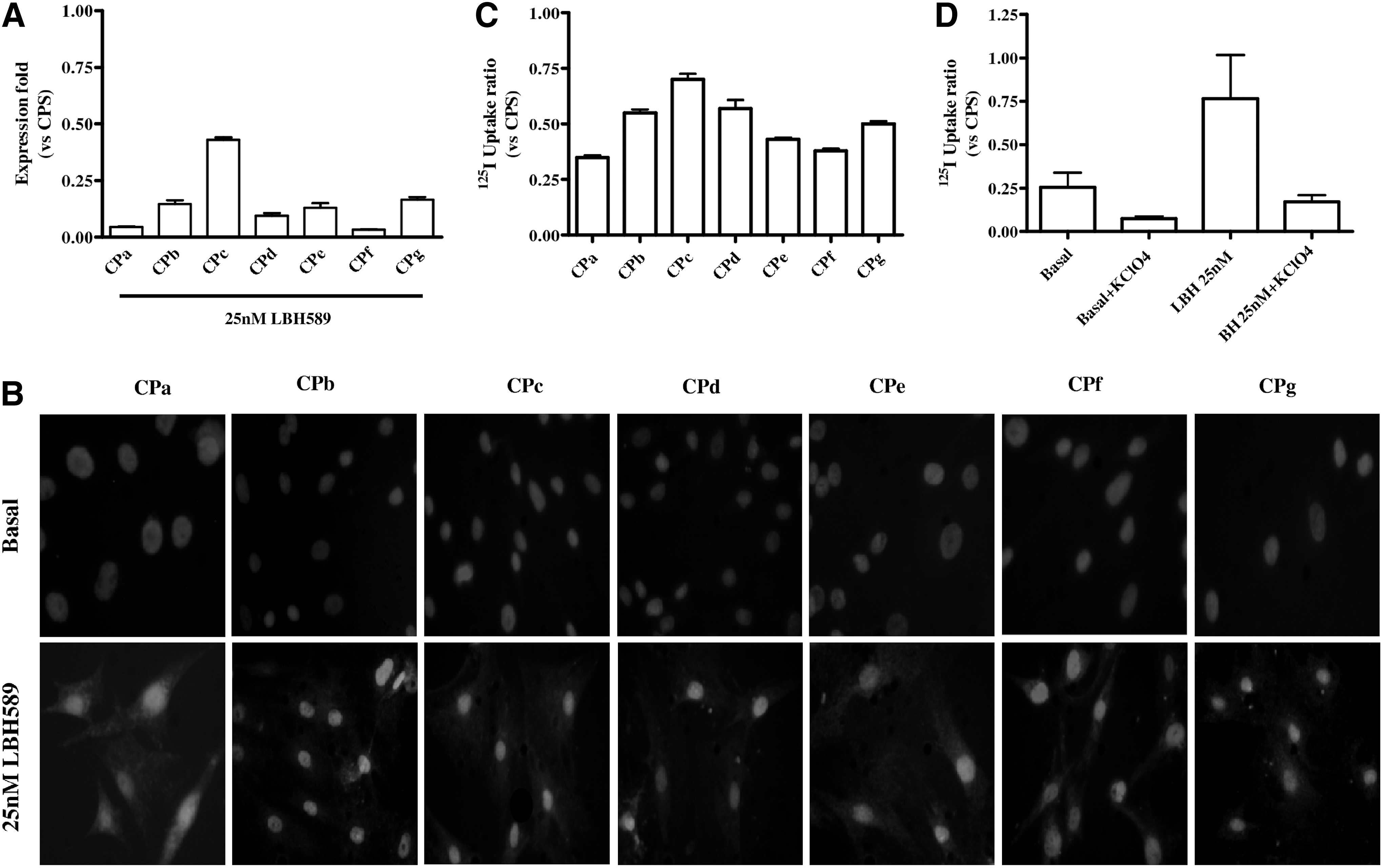
LBH589 effect on NIS gene expression in primary cultures
NIS protein and function
Immunofluorescence was performed on both stabilized cell lines and primary cultures to assess NIS protein expression. As shown in Figure 3A and B, no NIS protein was present in either BHT-101 or CAL-62 under basal conditions, but treatment with LBH589 induced the expression of the protein. This result was evident in all the primary cultures as shown in Figure 2B. NIS was correctly localized also in membranes of tumors treated with 25 nM LBH589 (5/5); all control cases were negative (0/5). A representative control case (Fig. 4A and B) and a treated tumor (Fig. 4C and D) are shown in Figure 4.
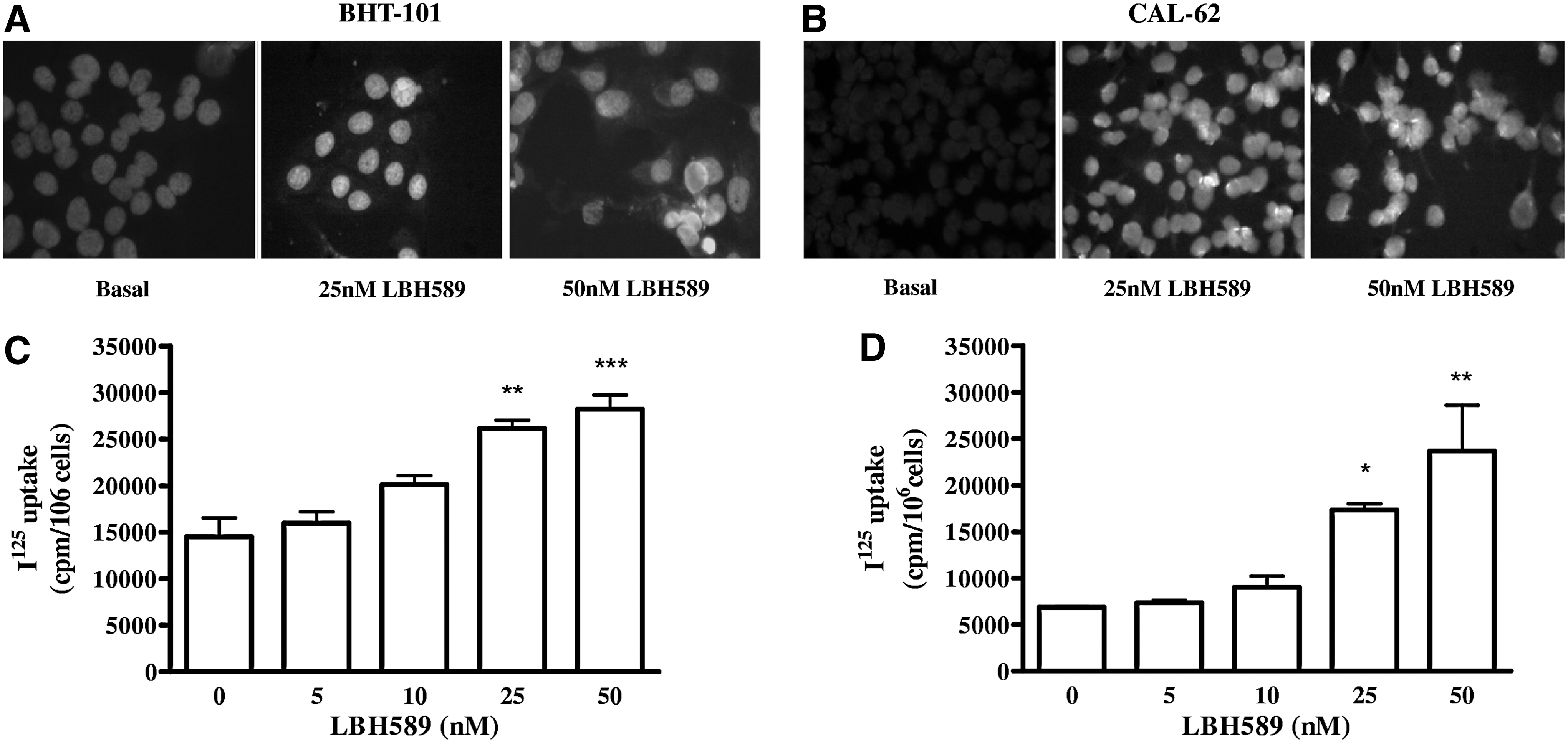
LBH589 effect on NIS protein expression in BHT-101
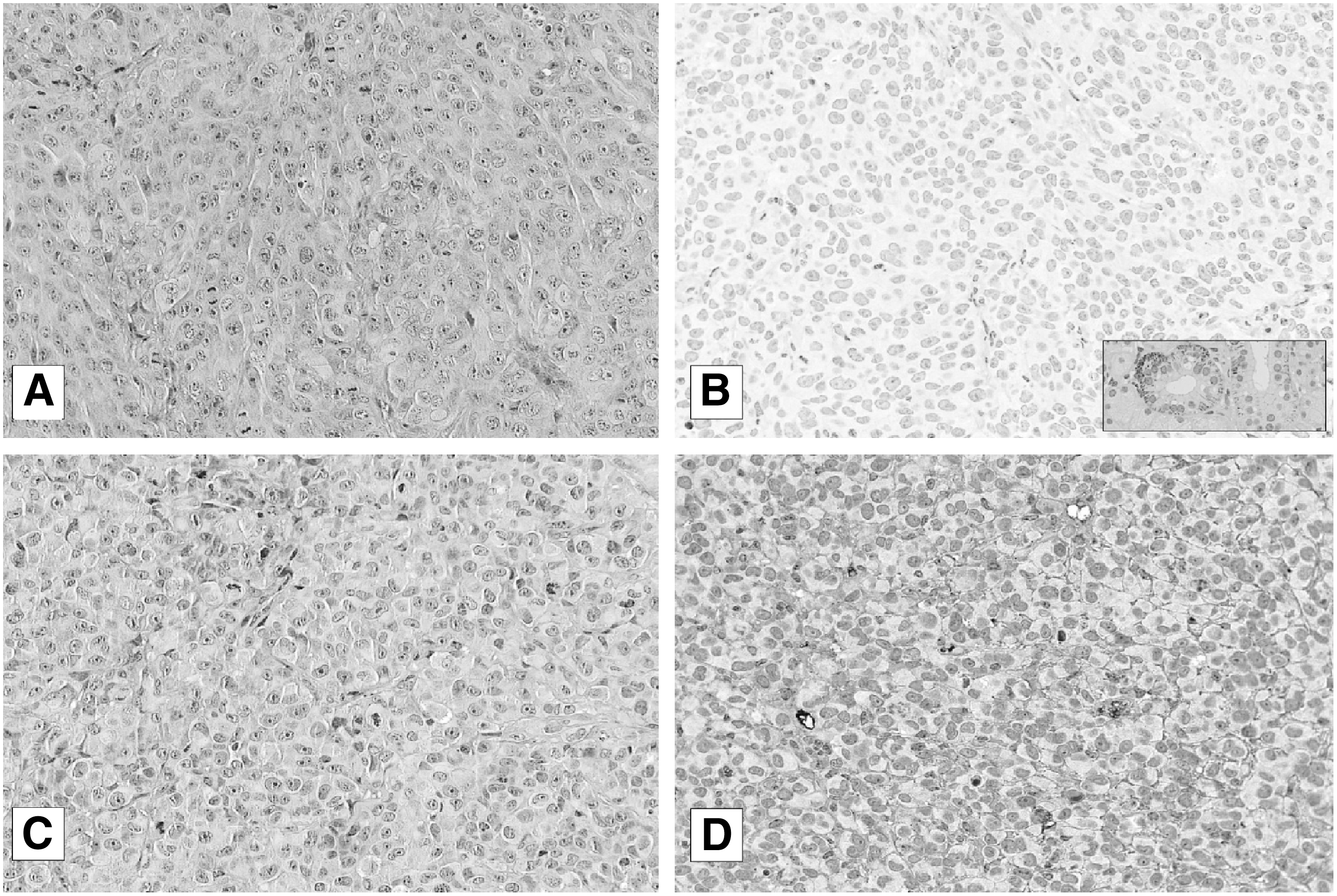
Immunohistochemistry (IHC) of NIS in a sample from an untreated tumor xenograft, hematoxylin and eosin (H&E)
125I uptake experiments confirmed not only the presence but also the function of the protein. In both stabilized cell lines we observed a dose-dependent increase of iodide uptake (Fig. 3C and D). In primary cultures, we chose a single concentration of 25 nM for the experiments of 125I uptake, and we found 125I uptake restoration after LBH589 treatment in every cell culture (Fig. 2C). Iodide uptake was specifically dependent on NIS because it was blocked in response to treatment with KClO4, as shown in Figure 2D. To evaluate the ability of ATC cells to retain iodide, we performed efflux studies, in which the cells were repeatedly incubated with HBSS. As reported in Figure 5, iodide retention was observed in both CAL-62 (83% retention after 5 min, t1/2=19 min) and CPa (79% retention after 5 min, t1/2=17 min), suggesting that iodide is adequately retained by ATC cells after treatment with 25 nM LBH589.
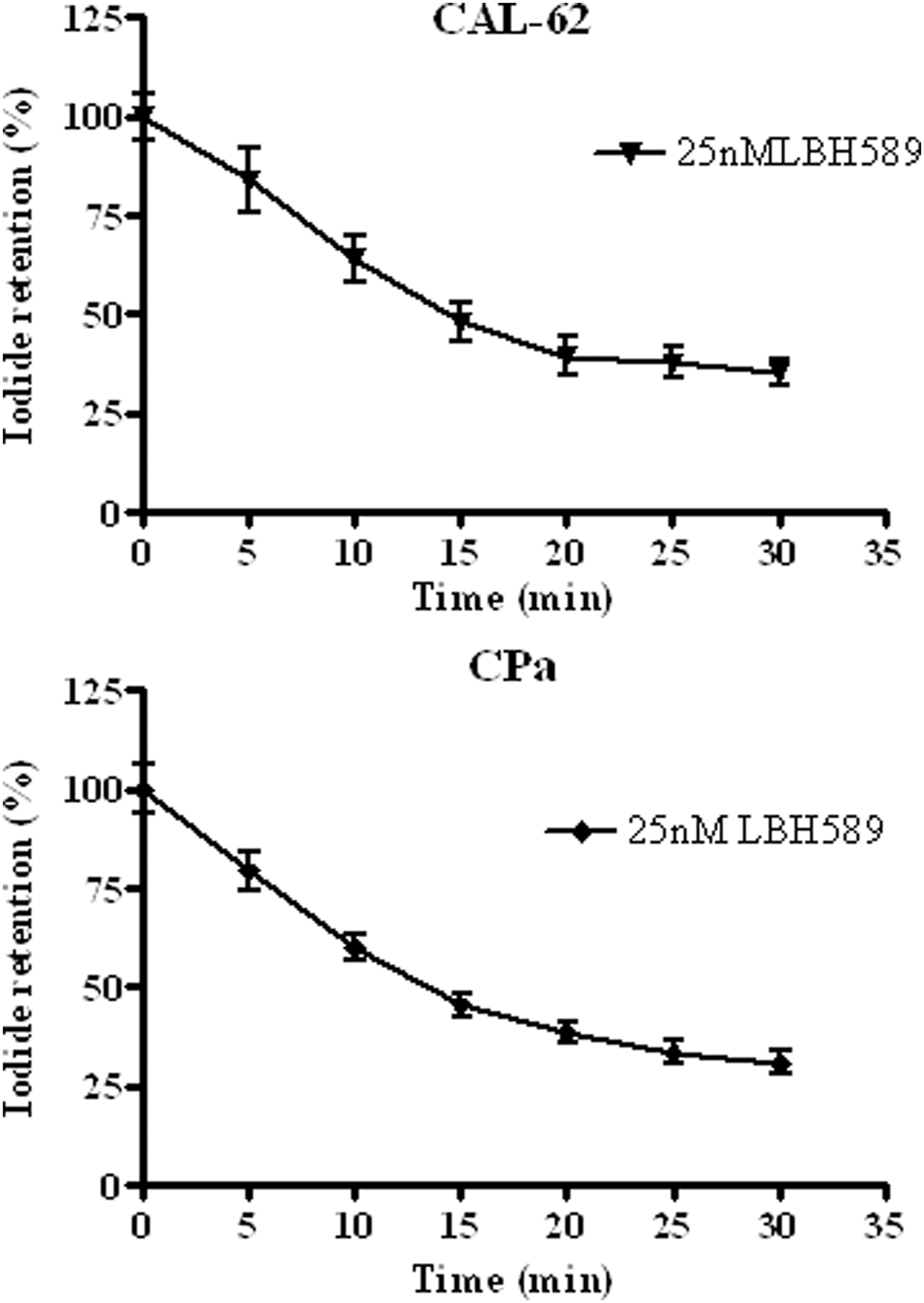
125Iodide retention in a stabilized cell line (CAL-62) and a primary culture (CPa) chosen as representative samples. Total radioactivity at the beginning of the efflux (100%) was calculated by adding the radioactivity of all media to those of the cells. The percentage of activity remaining in the cells at different times was obtained by subtracting the activity in each timed sample from that of the total radioactivity at the beginning. Results are normalized to total activity and cell number; experiments were performed in triplicate.
Iodide-induced cytotoxicity
Iodide uptake is not enough to deduce a possible cytotoxic effect of radioiodine on cells after LBH589 treatment. Therefore, to demonstrate cytotoxic radioiodine effects, a clonogenic assay with 131I was performed in a stabilized cell line (CAL-62) and in a primary cell culture (CPa). As shown in Figure 6, we observed a reduction in LBH589-treated colonies forming cells after 131I treatment in both cell lines; in fact, the number of colonies was significantly smaller in cells treated with 25 nM LBH589 compared to untreated controls (CAL-62: p<0.05; CPa p<0.01).
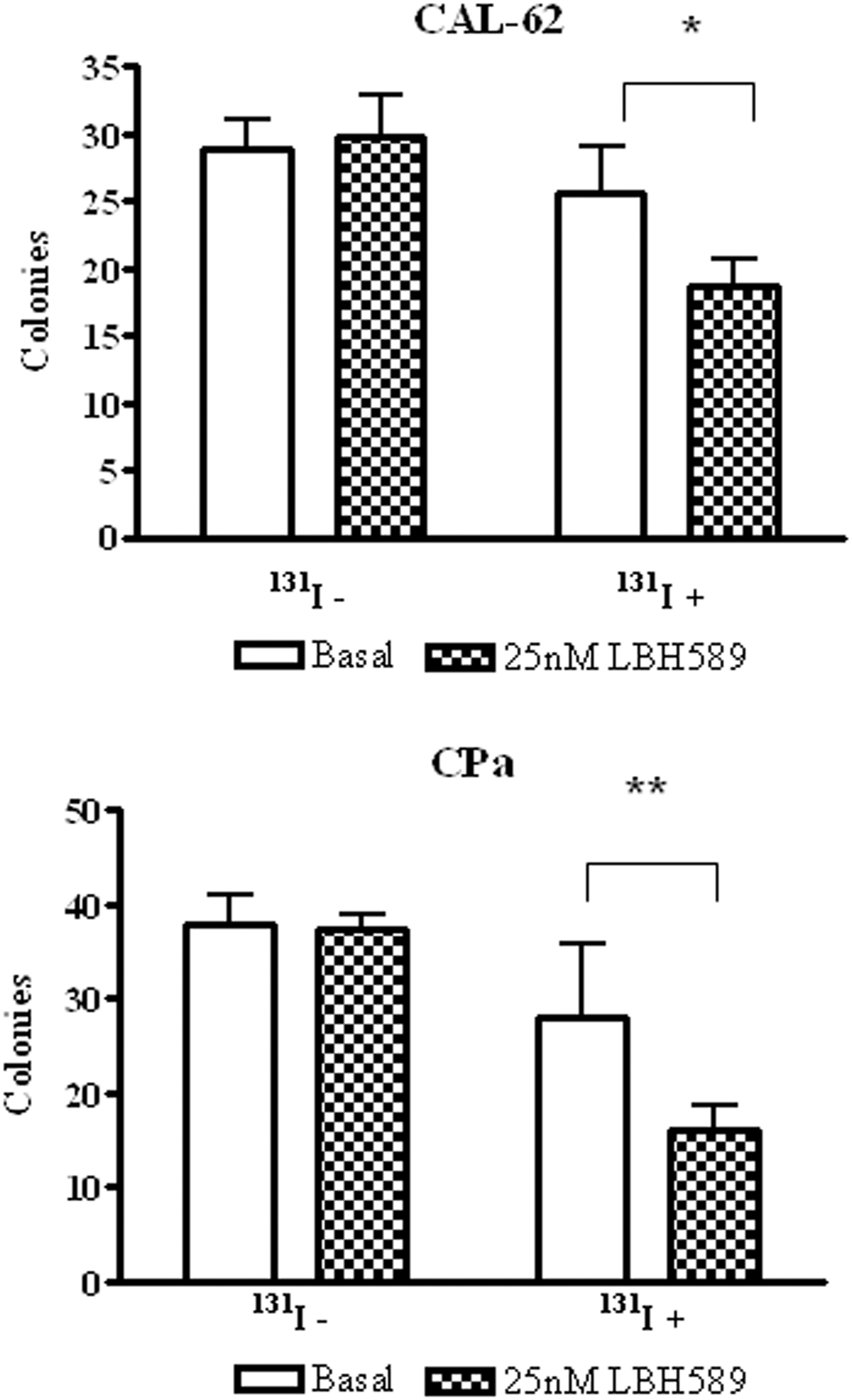
LBH589 effect on 131I cytotoxicity. 131I cytotoxicity was evaluated through a colony-forming assay as described; data are represented as number of colonies before (131I−) or after (131I+) the addition of 20 μM 131I to untreated cells (basal) or cells treated with 25 nM LBH589. Data are expressed as mean±SD; n=3. A representative stabilized cell line, CAL-62
Discussion
The absence of NIS, along with other iodide-handling proteins and specific thyroid transcription factors (18), characterizes ATCs and results in resistance to radioiodine treatment. Since epigenetic modifications contribute to the silencing of thyroid-specific genes in advanced cancers, drugs modifying the epigenome are considered a promising tool to obtain functional NIS (19). Recently it has been shown that global levels of acetylated histones are modified in thyroid cancer tissues and that lower levels of acetylated H3K18 are associated with tumor progression (20). In the present study we demonstrate that LBH589 induces NIS expression promoting the cytotoxic activity of radioiodine in ATC.
In fact, LBH589 induced NIS mRNA expression in a dose-dependent manner in both stabilized cell lines and in all the primary cultures obtained from patients who underwent thyroidectomy for ATC. The ability of other DCIs to act on NIS expression in ATC cells has already been reported (10,21,22). However, some previous studies showed the effect of DCIs in ARO and/or BHP cells (10,21), which are not of thyroid origin (23). The induction of NIS gene expression resulted in an increase in NIS protein expression, which was clearly demonstrated in all the cells we tested. In addition, in our in vivo model, NIS protein correctly localized in the membranes of tumors from LBH589-treated mice. Valproic acid as well as other drugs, such as 5′-aza-2’-deoxycytydine and retinoic acid were able to restore NIS mRNA and protein expression in thyroid cancer cell lines, but this treatment was unable to restore protein function (9,24).
Here we also show that LBH589 activated the NIS promoter in cells transfected with a construct containing the NIS promoter fused to luciferase. The NIS promoter can be activated by several thyroid-specific transcription factors (6,10). Even if various additional genes encoding tumor suppressors or proteins involved in thyroid hormone synthesis are epigenetically silenced in thyroid tumors (25,26), we could not find a significant modulation of the expression of the other studied genes (TTF-1, TG, PAX8, and TSH-R) in any of the cell lines, consistent with other studies (10). The observation that DCIs modulate NIS expression in nonthyroid cells (that are not expressing thyroid-restricted transcription factors) such as the human breast cancer cell line MCF-7 (10,11), strengthens the concept that LBH589 may directly affect the NIS promoter, even though the established mechanism has not been investigated.
We also demonstrate that LBH589 allowed iodide uptake and retention in both immortalized cultures of confirmed thyroid origin (23) and in primary cultures. The release of iodide that we observed was similar to that of cells stably expressing NIS (27,28). Our data finally demonstrate that LBH589 was effective in inducing cytotoxic effects mediated by radioiodine in ATC cells. The ability of LBH589 to express a functional symporter that allows radioiodine treatment appears extremely relevant and opens up therapeutic perspectives for undifferentiated thyroid tumors.
Because of its rarity and rapidly fatal course, ATC is difficult to investigate at both the clinical and the experimental levels (29). Nevertheless, testing drugs on primary cultures may be considered an interesting way to design new strategies for chemotherapy and gene therapy (30), with the aim of increasing the effectiveness of treatment in each single patient. We were able to set up primary cultures for studying the effects of LBH589. We constantly observed an increase in NIS expression, albeit at variable levels. These differences could be explained by a variable sensitivity to the drug due to the well-known heterogeneity of ATC. Moreover, the variability in iodide uptake in each primary culture is somehow similar to the great interpatient and even intrapatient spectrum in radioiodine kinetics that are observed in patients treated with standard 131I doses (31).
In conclusion, the present work suggests that the histone DCI LBH589 is an effective differentiating agent conferring radioiodine sensitivity to ATC cell lines and primary cultures. If confirmed, it provides a basis for clinical studies aimed at demonstrating improved radioiodine uptake in patients with ATC.
Footnotes
Acknowledgments
We thank Novartis Pharma AG (Basel, Switzerland) for providing us with LBH589, and Daniela Taverna and Francesca Orso (Dipartimento di Scienze Oncologiche, Universita` di Torino & M. B. C., Turin, Italy) for helping us with luciferase experiments. The study was supported by Fondazione CRT, Turin, and by Regione Piemonte Grants to Giuseppe Boccuzzi.
Author Disclosure Statement
The authors have nothing to disclose.