
Abstract
Background:
Maternal thyroid hormones play a fundamental role in appropriate fetal development during gestation. Offspring that have been gestated under maternal hypothyroidism suffer cognitive impairment. Thyroid hormone deficiency during gestation can significantly impact the central nervous system by altering the migration, differentiation, and function of neurons, oligodendrocytes, and astrocytes. Given that gestational hypothyroidism alters the immune cell ratio in offspring, it is possible that this condition could result in higher sensitivity for the development of autoimmune diseases.
Methods:
Adult mice gestated under hypothyroidism were induced with experimental autoimmune encephalomyelitis (EAE). Twenty-one days after EAE induction, the disease score, myelin content, immune cell infiltration, and oligodendrocyte death were evaluated.
Results:
We observed that mice gestated under hypothyroidism showed higher EAE scores after disease induction during adulthood compared to mice gestated in euthyroidism. In addition, spinal cord sections of mice gestated under hypothyroidism that suffered EAE in adulthood showed higher demyelination, CD4+ and CD8+ infiltration, and increased oligodendrocyte death.
Conclusions:
These results show for the first time that a deficiency in maternal thyroid hormones during gestation can influence the outcome of a central nervous system inflammatory disease, such as EAE, in their offspring. These data strongly support evaluating thyroid hormones in pregnant women and treating hypothyroidism during pregnancy to prevent increased susceptibility to inflammatory diseases in the central nervous system of offspring.
Introduction
T
In vivo and in vitro studies have identified most of the cellular and molecular targets of thyroid hormones in the CNS of offspring (10). In vivo studies have been performed by inducing hypothyroidism in pregnant animals (11). Neuronal migration, morphology, and synapsis are greatly affected in areas such as the cerebral cortex and hippocampus (12,13). The number of astrocytes (14) and oligodendrocytes (15) have also been shown to be altered in the progeny of thyroid-deficient mothers (16). In addition, the numbers of mature oligodendrocytes are reduced in the brains of progeny gestated by hypothyroid mothers, due to impaired proliferation and differentiation of oligodendrocyte precursor cells into mature oligodendrocytes (17). Furthermore, hypothyroidism during gestation decreases the number of myelinated axons and the thickness of the myelin sheath (18). Thus, there is extensive evidence indicating that gestational hypothyroidism can alter CNS development in the progeny in an irreversible manner, which supports the importance of diagnosing and treating this condition in pregnant women as stated by the most recent American Thyroid Association guidelines (8).
On the other hand, there is scarce information regarding the potential association between hypothyroidism and the susceptibility of the CNS to inflammatory damage by cells of the immune system. Few studies have analyzed the immune system of progeny gestated by mothers with hypothyroidism (19 –21) or progeny deficient in thyroid hormone receptor (22). However, the results show significant changes in thymus and spleen cellularity. Thyroid hormones can modulate expression of sonic hedgehog, which contributes to the regulation of thymic selection (23,24). Because malfunction of thymic selection can lead to the accumulation of autoimmune T cells, it is possible that the offspring of hypothyroid mothers could be more susceptible to autoimmune-inflammatory disorders (25). It is particularly important to determine whether hypothyroidism during gestation can increase CNS susceptibility to inflammatory-immune damage, such as is observed during multiple sclerosis (MS). MS is a chronic, neuroinflammatory demyelinating disorder of the CNS that predominantly affects young adults. There are approximately 2 million cases worldwide, affecting over 350,000 patients in North America (26). Because the etiology of most autoimmune-inflammatory disorders remains unknown, it is important to define whether gestational hypothyroidism could alter offspring susceptibility to inflammatory-autoimmune damage.
Here, we show that maternal gestational hypothyroidism significantly increased the severity of experimental autoimmune encephalomyelitis (EAE) in the offspring. EAE is an inflammatory disease of the CNS that has been widely used as a mouse model for MS (27 –29). In addition, to higher EAE disease scores, mice gestated under hypothyroidism showed an increased frequency of demyelinated spinal cord plaques, a higher number of CD4+ and CD8+ T cells, and a high percentage of death in oligodendrocytes in spinal cord sections. Our results highlight the importance of an adequate level of thyroid hormones during pregnancy to ensure resistance to CNS inflammation in offspring. Furthermore, our data reveal an unidentified link between the severity of CNS inflammatory disease in offspring and maternal thyroid hormone deficiency during gestation. These results support the importance of maternal thyroid hormone status for appropriate inflammation tolerance by the CNS of the progeny.
Materials and Methods
Mice
C57BL/6 mice were purchased from The Jackson Laboratory and maintained in a germ-free animal facility center at the Universidad Andrés Bello. All animal work was performed according to institutional and Comisión Nacional de Investigación Científica y Tecnológica de Chile bioethical guidelines, and were supervised by a veterinarian.
Induction of gestational hypothyroidism in mice
Gestational hypothyroidism was induced in pregnant mice using the following experimental design: Six- to eight-week-old female C57BL/6 mice were mated with C57BL/6 male mice. The day after mating, female mice were checked for vaginal plugs and a vaginal smear was obtained and analyzed under microscopy to search for spermatozoa. Mice with vaginal plugs or with a positive smear for spermatozoa were considered to be pregnant and that day was assigned as the first day of pregnancy (E1). To induce hypothyroidism, mice at E10 were treated with 0.02% 2-mercapto-1-methylimidazole (methimazole; MMI) in the drinking water until their last day of pregnancy. The untreated control group included pregnant mice that drank water without MMI during the entire pregnancy. A third experimental group consisted of pregnant mice that received MMI and T4 in the drinking water from E10 to the last day of pregnancy.
Detection of thyroid hormones and TSH
Thyroid hormones and TSH of mice in the three different experimental groups and their respective progeny were measured on the last day of pregnancy and at postnatal day 55. Blood (500 μL) was obtained from the tail of MMI, MMI+T4, and control pregnant mice, as well as from the offspring of the three experimental groups at postnatal day 55. Serum was separated from blood and used to measure total T3 (tT3) and total T4 (tT4) using radioimmunoassay and Coat-A-Count Siemens Healthcare Diagnostics kits (catalog no. TKT41 for tT3 and TKT31 for tT4). TSH was measured from serum using a mouse Ultrasensitive TSH enzyme-linked immunosorbent assay kit according to the manufacturer's instructions (MyBioSource; catalog no. MBS704901).
EAE induction and assessment
EAE was induced in the offspring of mice gestated in Hypo, Hypo+T4, and control mothers at the age of 7 weeks. The induction of EAE consisted of a subcutaneous injection in the flank of 50 μg myelin oligodendrocyte glycoprotein (MOG)–derived peptide (MOG35–55, sequence MEVGWYRSPFSRVV HLYRNGK) emulsified in complete Freund's adjuvant supplemented with heat-inactivated Mycobacterium tuberculosis H37 RA. These mice then received an intraperitoneal injection of 350 ng pertussis toxin, followed by a second intraperitoneal injection of 350 ng pertussis toxin 48 hours after the first injection. The day of the MOG injection was assigned as day 1. On this day, the disease scores of all mice were derived according to standard reported score criteria (30). Twenty-one days after MOG injection, mice were sacrificed for experimental analyses. In addition, a subset of animals was monitored for an extended period (35 days) for disease score evaluation.
Histological analyses
Histological analyses were performed to analyze myelin and cell infiltration in the spinal cord of female mice. In brief, mice were anesthetized with a mixture of 150 mg ketamine and 10 mg xylazine per kg of weight 21 days after EAE induction. Next, the mice were perfused through the heart with 30 mL 0.9% NaCl followed by 30 mL 4% paraformaldehyde (PFA) in phosphate-buffered saline (PBS). Spinal cords were isolated and the lumbar regions were dissected and carefully processed for histological analyses (n=6). Tissues were fixed for 1 hour in 4% PFA, dehydrated with ethanol, and embedded in paraffin. Sections (8 μm thick) were obtained using a microtome. Slices were stained with hematoxylin and eosin to analyze infiltrating cells or with Luxol Fast Blue to analyze demyelination. Histopathology examinations were performed in a blinded fashion to analyze both demyelination or cell infiltration. The scale used to evaluate inflammation and demyelination has been previously described (31,32) (see Supplementary Methods; Supplementary Data are available online at
Immunohistochemistry
Female mice were decapitated, and the spinal cord was removed and fixed in 4% PFA. The spinal cord was frozen in OCT with isopentane in liquid nitrogen. Lumbar sections (20 μm thick) were obtained using a cryostat (Leica CM152S) and the tissue sections were fixed in 4% PFA. The tissues were immunostained with 3 μg/mL antimouse myelin basic protein (MBP; Abcam). Antimouse Alexa 594 (Invitrogen) was used as secondary antibody (final concentration of 10 μg/mL). Nuclei were counterstained with Hoechst. To assess inflammatory infiltration, tissues were immunostained with 5 μg/mL of the anti-CD4 Alexa 488–conjugated mouse antibody (Invitrogen; catalog number MCD0420) that was mixed with 5 μg/mL of anti-CD8 rat antibody (Abcam; catalog number Ab22378). The secondary antibody to detect anti-IgG rat was an Alexa 647–conjugated goat antibody (Invitrogen; catalog number A21247) at a final concentration of 10 μg/mL. Nuclei were counterstained with Hoechst. Immunofluorescence was analyzed using a Fluoview FV1000 laser scanning confocal microscope (Olympus) with a 20× and 60× objective. CD4- or CD8-positive T cells were quantified in a blind fashion from three independent experiments. Three lumbar sections per mouse were analyzed. In each lumbar section, three different areas were chosen to quantify the total number of cells (Hoechst staining) as well as CD4- or CD8-positive cells. Positive cells for CD4 or CD8 were expressed as the percentage of total cells.
Western blot
Twenty-one days post-EAE induction, spinal cords from the offspring of every experimental group were isolated, homogenized, treated with lysis buffer (20 mM HEPES, 0.4 M NaCl, 5 mM MgCl2, 0.5 mM EDTA, 0.1 mM phenylmethanesulfonyl fluoride, 1 μM leupeptin, 0.1 mM NaF, 0.5 mM o-Vanadate, and 1 mM aprotinin), and total protein was determined in these samples. Samples were resolved using 12.5% sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes (Thermo Scientific). After blocking with 5% nonfat milk in 0.1% Tris-PBS, membranes were probed overnight at 4°C with a 1:300 dilution of mouse anti-MBP (Abcam) and a 1:3000 dilution of anti-β-actin (A-5441; Sigma-Aldrich) in 0.1% Tris-PBS. A horseradish peroxidase–conjugated antimouse antibody was used as the secondary antibody, and proteins were visualized by chemiluminescence (Amersham, GE Healthcare).
Oligodendrocyte death analyses
Death of oligodendrocytes in female spinal cord lumbar sections was analyzed using terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining. Staining was performed according to manufacturer's instructions using the in situ Cell Death Detection kit (Roche Applied Science). Briefly, mice were decapitated and the spinal cord was removed, fixed in 4% PFA, and frozen in OCT with isopentane in liquid nitrogen. Lumbar sections (10 μm thick) were obtained using a cryostat (Leica CM152S) and were fixed in 4% PFA. The tissues were incubated with the TUNEL reaction mixture kit. To label mature oligodendrocytes within these tissues, immunofluorescence analyses were performed using a polyclonal antibody against 2′-3′-cyclic nucleotide 3′-phosphodiesterase (CNPase). Spinal cord sections were immunostained with 10 μg/mL rabbit CNPase antibody (Sigma; catalog number C9794). A secondary antirabbit Cy-5 antibody (Chemicon; catalog number AP132S) was used at a final concentration of 15 μg/mL. Nuclei were counterstained with Hoechst. Immunofluorescence was analyzed using a laser scanning confocal microscope (Fluoview FV1000; Olympus) with a 20× objective. CNPase and TUNEL quantification was performed using ImageJ image analysis software (NIH; see Supplementary Methods). Dead oligodendrocytes were quantified in three independent experiments by double blinded analyses. For each experiment, pictures were taken for three lumbar sections of one mouse. Dead oligodendrocytes were quantified in the field by counting the total number of cells co-stained for CNPase and Hoechst using a grid per slice method. The percentage of dead oligodendrocytes was calculated by counting cells that were positive for CNPase, TUNEL, and Hoechst. This number was divided by the total number of CNPase-positive cells in the same grid. The results are expressed as the percentage of total oligodendrocytes that were positive for TUNEL.
Statistical analyses
Data and statistical analyses were performed using Prism 4 software (Graph Pad Software, Inc.) and Statistica 6.0 software (StatSoft Inc., 2001). Statistical differences for thyroid hormones and TSH were tested using unpaired Student's t-tests. The marble burying test and TUNEL experiments were analyzed using a factorial analysis of variance (ANOVA) test with fixed factors. Finally, we used an ANOVA test with repeat measurements to analyze the effect of gestational hypothyroidism on EAE clinical scores. To assess oligodendrocyte death, data are expressed as mean±standard error and comparisons were performed using the two-tailed Student's test; statistical significance is indicated by *p<0.0001.
All data were tested for normality and homoscedasticity using Kolmogorov–Smirnov and Levene's tests. When necessary, data were transformed to meet statistical assumptions. When differences were significant at p<0.05 after general linear model tests, we used a posteriori Bonferroni test for multiple comparisons and Tukey's test for balanced samples.
Results
Induction of gestational hypothyroidism
To induce gestational hypothyroidism, a group of pregnant mice were treated with the thyroperoxidase inhibitor MMI in the drinking water during days 12–21 of pregnancy (E12–E21) (33). This group was designated as MMI. To determine whether our observations were due to the low levels of thyroid hormone and not to the side effects of MMI, we included a second experimental group of pregnant mice that were provided water supplemented with MMI and T4 (designated as MMI+T4) from E12 to E21 (34). The control group was composed of euthyroid pregnant mice that drank water with neither MMI nor T4 during the entire pregnancy. To corroborate that we induced gestational hypothyroidism, serum levels of tT4, tT3, and TSH were measured at the end of pregnancy in all experimental groups. Figure 1A shows that tT4 was significantly reduced in MMI-treated pregnant mice (1.54±0.45 μg/dL) compared to control pregnant mice (3.78±0.24 μg/dL) and MMI+T4-treated pregnant mice (4.23±0.28 μg/dL). There were no significant differences in tT4 within the control group versus the MMI+T4-treated group (Fig. 1A). The levels of tT3 followed the same pattern as tT4 (Fig. 1A). tT3 was significantly reduced in MMI-treated pregnant mice (30.9±2.78 ng/dL) compared to control pregnant mice (84.33±9.47 ng/dL) and MMI+T4-treated pregnant mice (88.40±1.66 ng/dL). There were no significant differences in tT3 between the control group and MMI+T4-treated mice (Fig. 1A). The TSH level was significantly higher in MMI-treated pregnant mice (2.72±0.133 μIU/dL) compared to control animals (1.19±0.09 μIU/dL) and MMI+T4-treated pregnant mice (1.27±0.09 μIU/dL; Fig. 1A). There were no significant differences in TSH levels between the control group and the MMI+T4-treated group (Fig. 1A). Analyses of these results support the idea that MMI treatment successfully induced hypothyroidism in pregnant mice, and that treatment with T4 led to recovery of the euthyroid condition in pregnant mice treated with MMI.
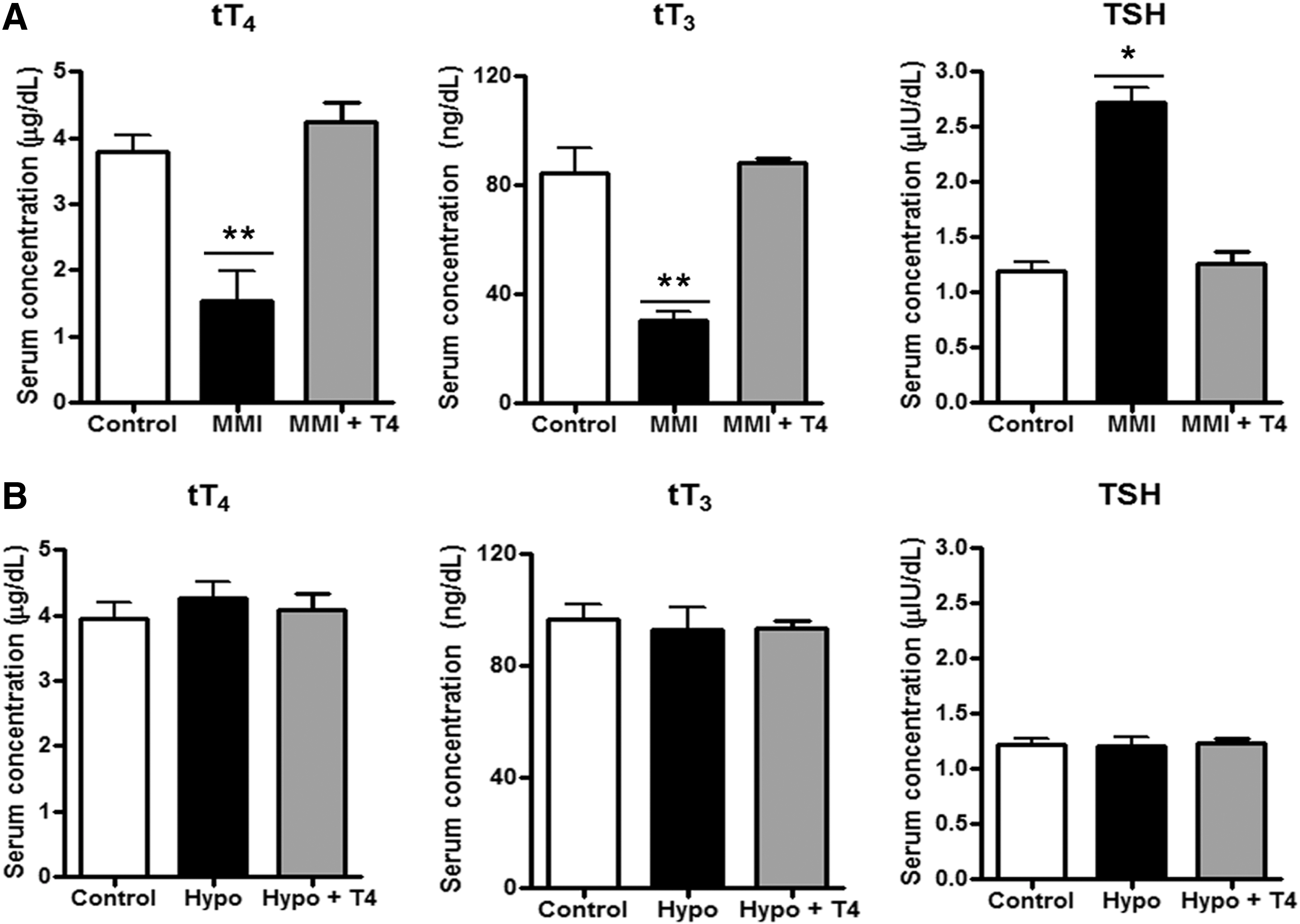
MMI treatment induces hypothyroidism in pregnant mice.
It has been reported that progeny gestated under hypothyroid conditions are born prematurely compared to mice gestated under euthyroid conditions (10,35). Supplementary Figure S1A shows that progeny gestated in hypothyroidism (hereafter referred to as Hypo offspring) were born prematurely compared to progeny gestated under euthyroid conditions (hereafter referred to as control offspring) or progeny gestated in mice treated with MMI+T4 during pregnancy (hereafter referred to as Hypo+T4 offspring). Gestation periods for the control and Hypo+T4 progenies were similar, indicating that the addition of T4 to MMI-treated mice during gestation recovered euthyroid pregnancy condition. We also tested hippocampal function in these three progeny groups because it has been shown that gestational hypothyroidism induces deterioration of the hippocampus (36). Progeny were subjected to the marble-burying test, a behavior test used to determine hippocampal function (37). Representative images from before and after the test are shown in Supplementary Figure S1B: images on the left show the initial marble distribution (time 0); images on the right were obtained after mice had spent 30 minutes in the cage with the marbles. The percentage of marbles that were moved or buried were quantified and plotted in Supplementary Figure S1C. We observed that Hypo offspring showed less marble burying activity than control and Hypo+T4 offspring (Supplementary Fig. S1B, C). These results support the notion that Hypo offspring have altered hippocampal function and that T4 treatment during gestation protects against hippocampal damage induced by MMI. To discard the possibility that MMI treatment during gestation can imprint a thyroid hormone imbalance in adulthood of the Hypo progeny, thyroid hormones and TSH were measured in the progeny (Fig. 1B). Analyses of these results showed that tT4, tT3, and TSH levels were normal in control, Hypo, and Hypo+T4 offspring, thus indicating that all progeny were euthyroid in adulthood.
Gestational hypothyroidism increases EAE severity in female offspring
EAE was induced in adult Hypo, control, and Hypo+T4 offspring. We determined the percentage of mice in each experimental group that acquired EAE and recovered from this disease. Hypo and control offspring had an equivalent susceptibility to EAE (Supplementary Table S1). However, only 20% of Hypo progeny recovered from EAE compared to 32% of control progeny (Supplementary Table S1). These results suggest that hypothyroidism during pregnancy increases the risk of progeny suffering a more severe form of EAE. To analyze the intensity of EAE, the disease score of EAE for each progeny was plotted in Figure 2. The disease score was significantly higher for Hypo offspring compared to control or Hypo+T4 offspring. However, the disease score of control and Hypo+T4 offspring was similar. Analyses of these results support that offspring gestated under hypothyroid conditions suffer from more severe EAE compared to offspring gestated under euthyroid conditions. Interestingly, when we plotted the EAE disease score separately for each sex, we observed than only female Hypo offspring had a significantly higher EAE disease score compared to control or Hypo+T4 offspring (Supplementary Fig. S2A). Although the EAE disease score for male Hypo offspring tended to be higher, it was not statistically different compared to control or Hypo+T4 offspring (Supplementary Fig. S2B). To evaluate the progression of EAE in control and Hypo EAE, the EAE disease score was evaluated until 35 days post-EAE induction (Supplementary Fig. S2C). These results indicate that both progeny reached a plateau in the EAE score 29 days after EAE induction. Because we found that the disease score was significantly higher only for Hypo female offspring compared to control or Hypo+T4 offspring, the following experiments in this work were conducted only with female progeny.
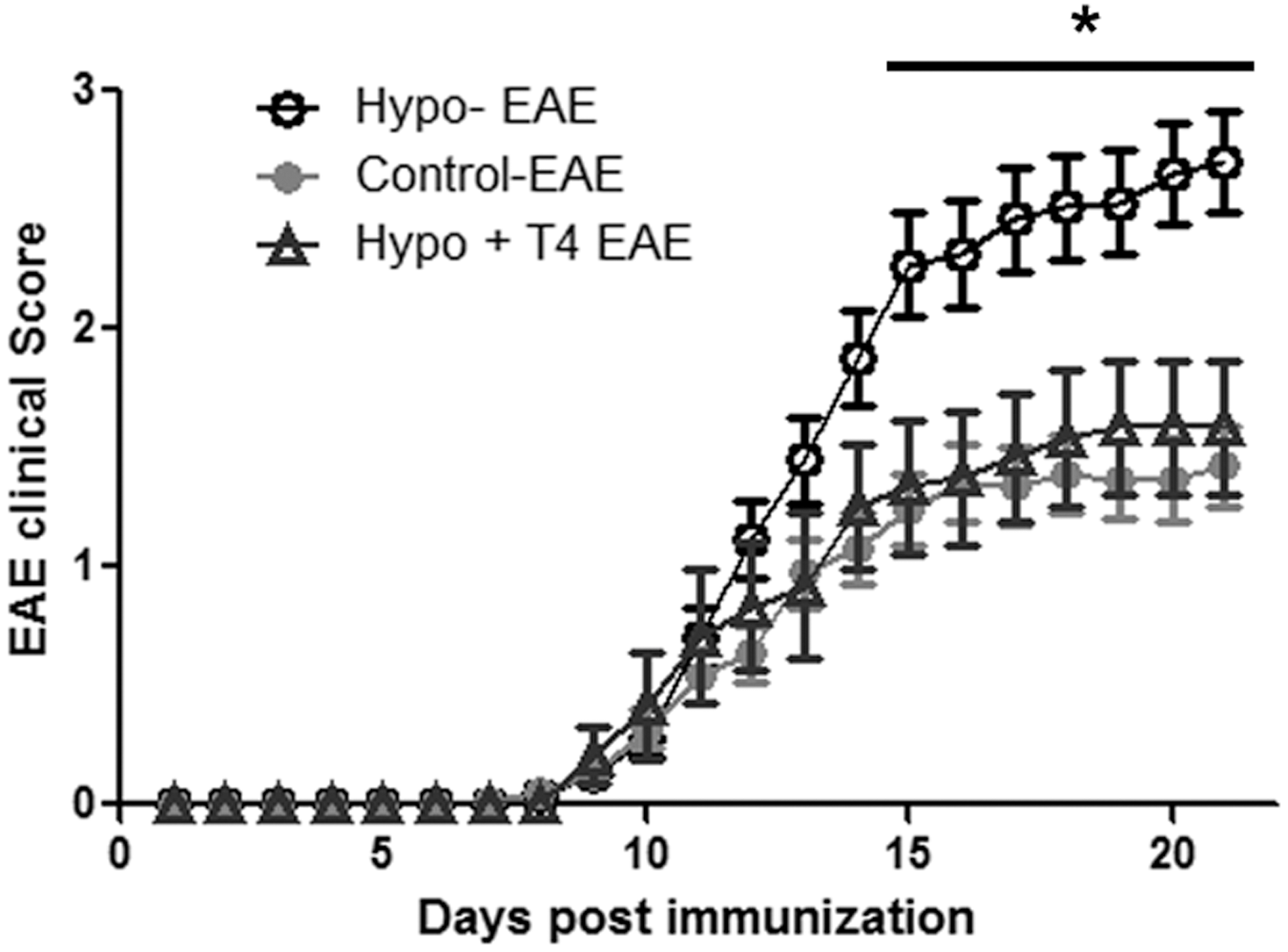
Higher EAE disease scores in mice gestated in hypothyroid mothers. Disease EAE scores were recorded for each mouse after EAE was induced until day 21. EAE induction was done at day 0. The graph shows the mean of the disease scores for the female and male offspring±standard deviation for those gestated under Hypo (◯, n=52), control (●, n=51), or Hypo+T4 (△, n=12) conditions. *p<0.05. EAE, experimental autoimmune encephalomyelitis.
Demyelination is increased in the spinal cord of female offspring that have EAE and were gestated in hypothyroid conditions
Spinal cord sections from female control, Hypo, and Hypo+T4 offspring induced with EAE were analyzed for myelin by staining with Luxol Fast Blue. Figure 3A shows representative pictures of these analyses, and Figure 3B shows quantitative analyses of the Luxol fast blue staining. Higher demyelination was observed in Hypo offspring. The myelination areas have cell infiltration (see pictures with higher magnification in Supplementary Fig. S3A). The analyses of these results indicate that Hypo female offspring had less myelin staining than control or Hypo+T4 offspring. These results were corroborated by the detection of MBP immunofluorescence in spinal cord sections from these offspring (Fig. 3C) and using Western blot analyses for MBP (Fig. 3D). We found reduced MBP staining caused by EAE in control, Hypo, and Hypo+T4 offspring compared to the respective control offspring without EAE (Fig. 3C). Pictures with higher magnification are found in Supplementary Figure S3B. These findings were supported by MBP Western blots analyses (Fig. 3D). These data also correlate with fluoromyelin staining (Supplementary Fig. S3C) and Western blot analysis of CNPase (Supplementary Fig. S3D) (31,32). Each of these approaches support the notion that mice with EAE that were gestated under hypothyroid conditions have higher spinal cord demyelination. These results are in agreement with the severity observed in the disease score data (Fig. 2), less myelin staining, and the MBP and CMPase content that was observed in the white matter region from the spinal cords of Hypo EAE-suffering female offspring compared to female offspring that were gestated in euthyroid conditions (both control and Hypo+T4 offspring).
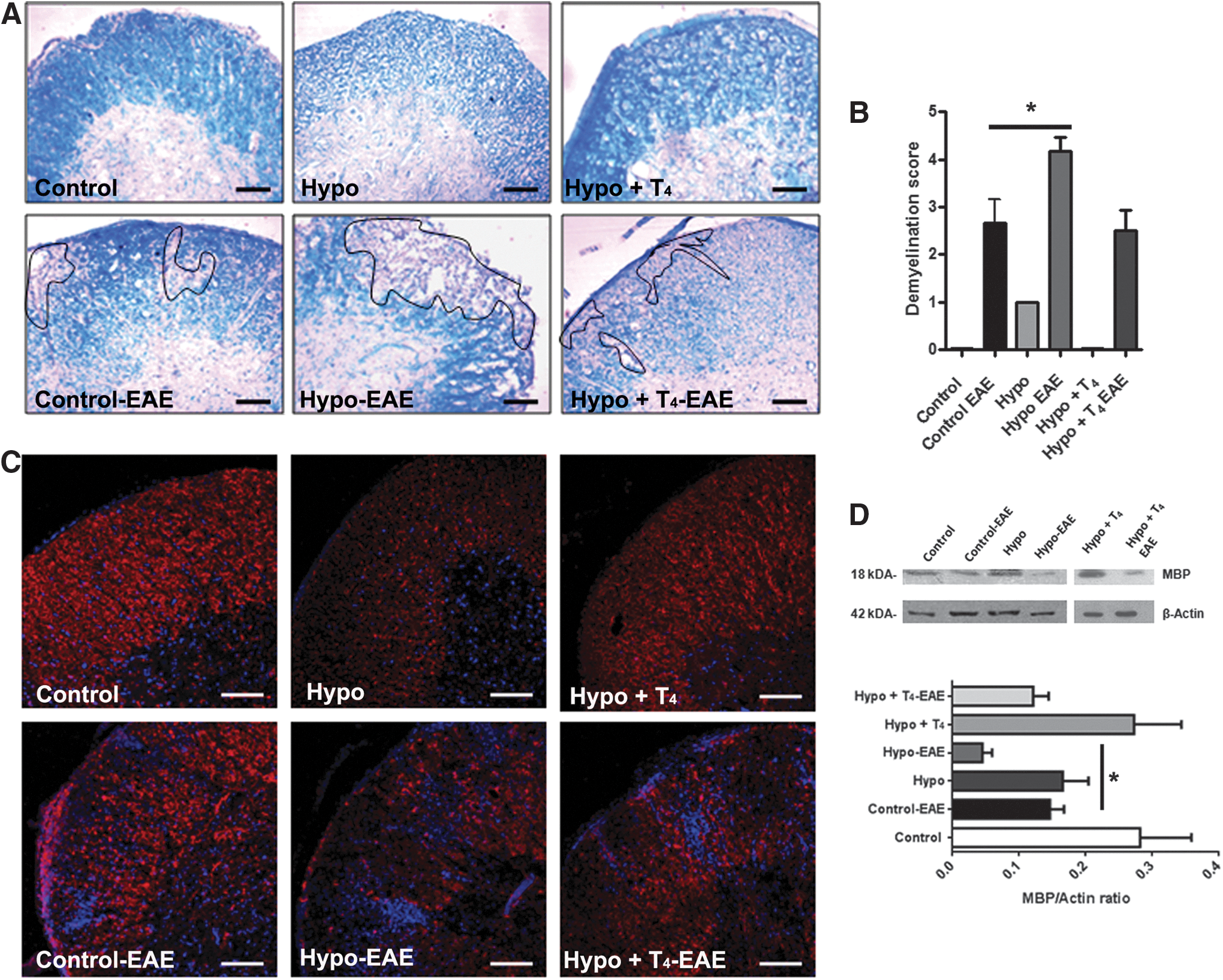
Gestation under hypothyroid conditions increases demyelination in the spinal cord of mice suffering from EAE.
Inflammatory cell infiltration is increased in the spinal cord of female offspring that have EAE and were gestated under hypothyroid conditions
Spinal cord sections of female control, Hypo, and Hypo+T4 offspring induced with EAE were stained with hematoxylin and eosin (Fig. 4A). Pictures with higher magnification are in Supplementary Figure S4A. Infiltration was quantified using a pathologic score (Fig. 4B) (31,32). Analyses of these results indicate that although infiltrating cells due to EAE were detected in control, Hypo, and Hypo+T4 female offspring, the infiltration was significantly higher in Hypo female offspring (Fig. 4A, B). To determine that the cell infiltration corresponds to T cells, immunofluorescence analyses were performed to detect CD4+ and CD8+ cells in the spinal cord sections of all experimental female offspring groups induced with EAE (Fig. 4C). Pictures with higher magnification are in Supplementary Figure S4B. Quantitative analyses of CD4+ and CD8+ were plotted in Figure 4D. These results indicate that Hypo female offspring suffering EAE have a significantly higher number of CD4+ and CD8+ cells in the spinal cord compared to all other experimental groups, and indicate that offspring with EAE have increased spinal cord inflammation if they are gestated under hypothyroid conditions.
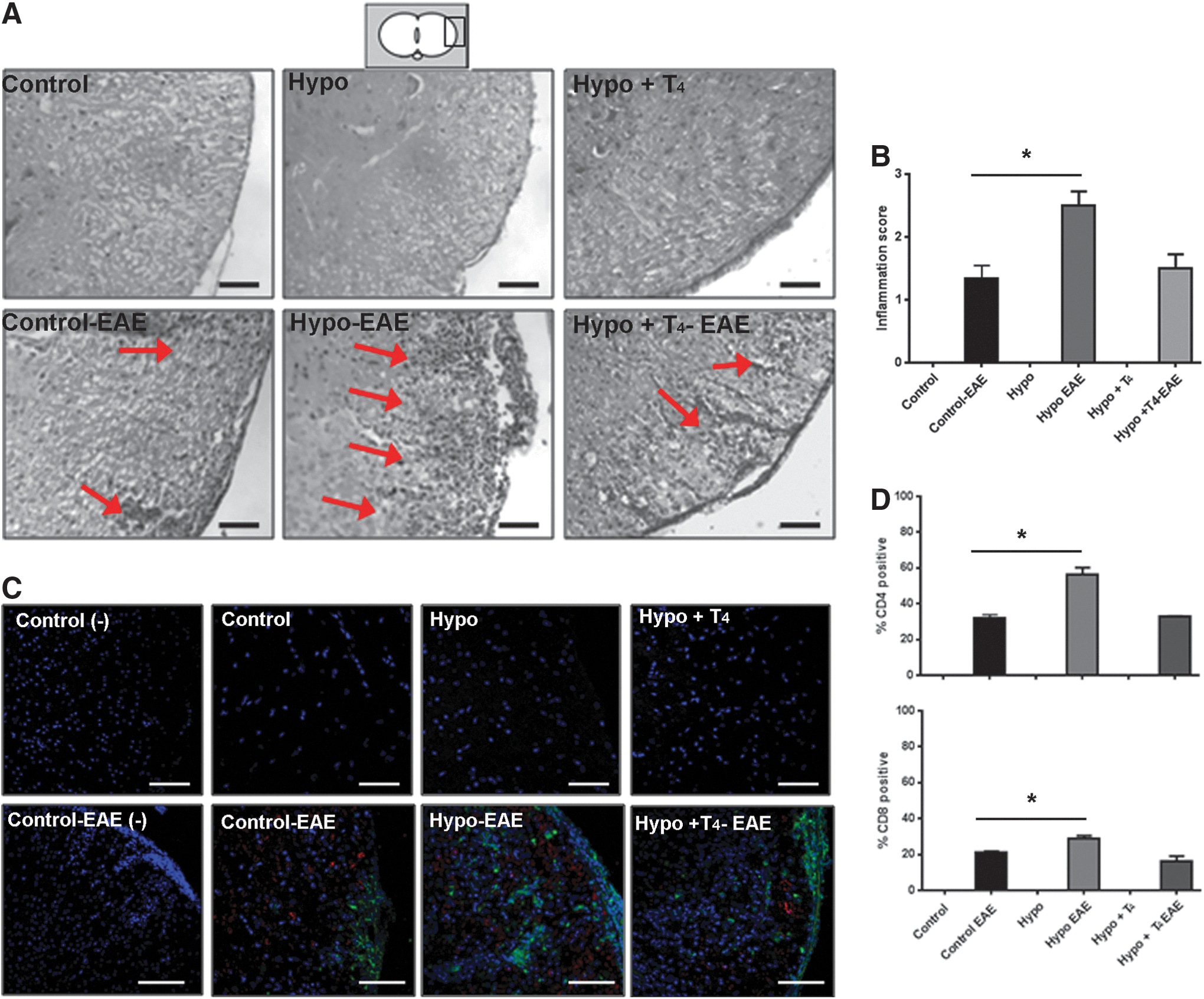
Gestation under hypothyroid conditions increases inflammatory infiltrates in the spinal cord of mice suffering from EAE.
Female mice gestated in hypothyroid mothers show increased oligodendrocyte death in spinal cord
To analyze oligodendrocyte death from spinal cord sections of control, Hypo, and Hypo+T4 female offspring with EAE, tissues were labeled with TUNEL (green fluorescence) and co-stained with anti-CNPase antibodies, an oligodendrocyte marker (orange fluorescence) (Fig. 5A). TUNEL- and CNPase-positive cells were quantitated and their percentages were plotted (Fig. 5B). These results indicate that TUNEL-positive oligodendrocytes were detected in all offspring suffering from EAE. Hypo female offspring with EAE showed significantly more TUNEL-positive cells than control or Hypo+T4 offspring (Fig. 5A, B). Hypo+T4 offspring showed no significant differences compared to the control group. These results suggest that offspring gestated under hypothyroid conditions have an impaired capacity to regulate CNS inflammation when have EAE compared to offspring gestated under euthyroid conditions.
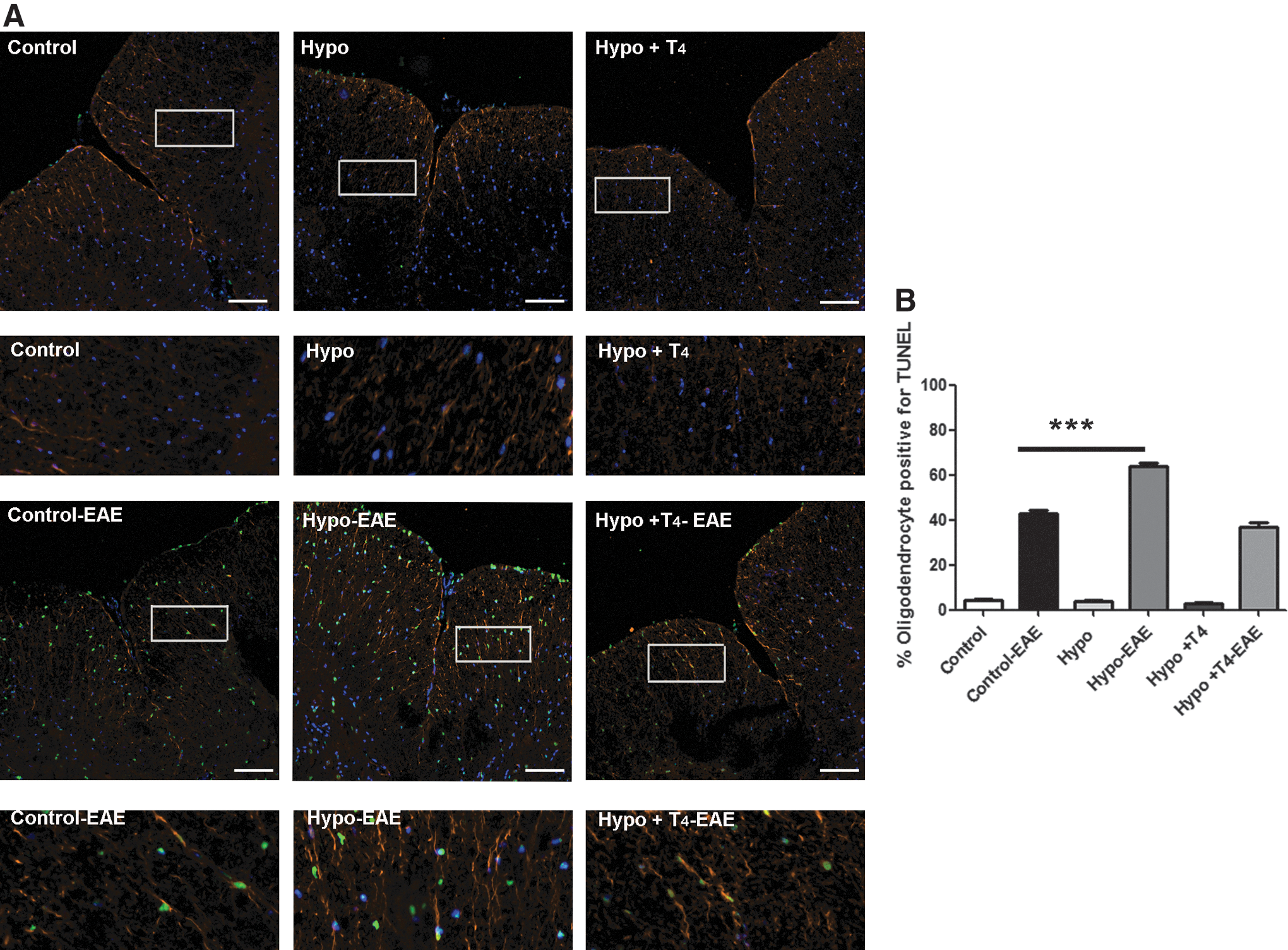
Gestation under hypothyroid conditions increases oligodendrocyte death in the CNS of offspring suffering from EAE.
Discussion
Here we demonstrate for the first time that gestational hypothyroidism imprints offspring with the potential for suffering a worse course of EAE compared to gestation under euthyroid conditions. EAE severity of Hypo offspring was reverted when T4 was added during gestation, indicating that the cause of higher EAE scores in these offspring was due to thyroid hormone deficiency during gestation rather than a side effect of MMI. In addition, the fact that Hypo offspring were euthyroid in adulthood supports the finding that the higher EAE clinical scores in this group is due to a thyroid hormone deficiency during pregnancy and not thyroid hormone alterations in adulthood. Although TSH and thyroid hormone levels are normal in the adult offspring, expression of their thyroid hormones receptors could be altered. Kilby et al. showed that thyroid hormone deficiency during gestation significantly reduced thyroid hormone receptor expression in human fetal cerebral cortex and cerebellum (38). However, there are no reports suggesting that the reduction of thyroid hormone receptors is maintained until adulthood.
We found that Hypo male offspring have a tendency of higher EAE clinical scores, but only Hypo female offspring have significant differences compared to controls. In fact, women have a higher incidence of MS compared to men (39). It has been shown that estrogen immunosuppresses and delays the appearance of EAE (40) and a reduction in estrogen increases production of inflammatory cytokines such as IL-6 and tumor necrosis factor (41). Moreover, estrogen enhances re-myelination and increases the thickness of myelin sheaths (42). Therefore, our finding that female offspring have more severe EAE could be because gestational hypothyroidism affects estrogen production in adulthood. We have not found any published reports regarding the impact of gestational hypothyroidism on estrogen production in the adult offspring. Currently, it only is known that thyroid hormones are important for the estrous cycle, and hypothyroid rats have been shown to have an alteration in their cycle that is recovered when T4 is administered (43).
Despite the fact that Hypo offspring suffered more severe EAE than mice gestated under euthyroid conditions, they did not have significant differences in the frequency of EAE occurrence, suggesting that the susceptibility to EAE does not increase with gestational hypothyroidism. On the other hand, we found that only 20% of mice gestated under hypothyroid conditions recovered from EAE compared to 32% of mice gestated under euthyroid conditions (Supplementary Table S1). These data suggest that mice gestated under hypothyroid conditions have a reduced capacity to modulate the damage of an inflammatory response in the CNS. Disease score data (Fig. 2) were consistent with strong spinal cord demyelination (Fig. 3) and inflammation (Fig. 4) shown by the Hypo-EAE offspring compared to control-EAE offspring. Importantly, this phenotype was reverted in the Hypo+T4 group, confirming that the lack of thyroid hormone during gestation is directly responsible for the increased severity of EAE in adulthood. We found that Hypo-EAE offspring had lower MBP (Fig. 3D), lower CNPase expression (Supplementary Fig. S3B), and less staining for myelin lipids (Fig. 3A and Supplementary Fig. S3A) than control-EAE offspring. Even though, our data show that Hypo mice have less MBP and CNPase than controls, the reduction of these proteins after EAE is greater in Hypo-EAE mice than in control-EAE mice. Furthermore, Hypo-EAE offspring have an increased number of oligodendrocytes that stain positive for TUNEL in the lumbar sections of the spinal cord (Fig. 5A, B). These results suggest that oligodendrocytes from Hypo+EAE offspring may be more susceptible to inflammation or that the inflammation developed in EAE is much stronger than that in control mice. In fact, we found more CD4- and CD8-positive cells in the spinal cord of Hypo-EAE mice compared to control-EAE (Fig. 4C, D), supporting the latter hypothesis. It may be possible that the immune response in mice gestated under hypothyroid conditions leads to exacerbated inflammatory damage compared to mice gestated in euthyroid mothers. Information is scarce regarding the immune system of offspring gestated under hypothyroid conditions. It has been reported that during the first postnatal weeks of exposure to propylthyouracil, a significant decrease in cell number occurs in the thymus and spleen (19). Although some reports have suggested an increase in the frequency of naive and regulatory T cells, (44) others have shown an increase in the activity of effector T cells (45). Furthermore, in T3-receptor–deficient mice, it was demonstrated that deficient thyroid hormone function causes an irreversible decrease in several immune cells, including B and T cells, macrophages, and granulocytes (22). In addition, the expression of sonic hedgehog, a molecule involved in thymic function (46), is modulated by thyroid hormones during embryogenesis (47). However, additional functional studies are required to understand the effect of gestational hypothyroidism on the actions of the immune system in the progeny. An alternative mechanism that may contribute to the phenotype of the mice suffering more severe EAE is an alteration in the permeability of the blood–brain barrier. Although the formation of the blood–brain barrier occurs postnatally (48), it is possible that gestational hypothyroidism could alter the progenitor cells that form the blood–brain barrier, such as endothelial cells and/or astrocytes, through an epigenetic mechanism (49).
Our results support an association between gestational hypothyroidism and inflammatory disease severity in adult progeny. These findings are highly relevant for the etiology of MS, which is known to be genetic in only 5% of cases. For the remaining 95% of patients with MS, the etiology is unknown (50). It is thought that environmental, infectious, and immunological factors can contribute to triggering MS in susceptible patients (51). Low levels of thyroid hormones during pregnancy could also contribute to creating an environment more susceptible to MS development or result in more severe symptoms of this disease.
Our data suggest that gestational hypothyroidism increases the likelihood of the offspring suffering from more severe inflammatory diseases in the CNS. These results could have a favorable impact for public health by promoting the obligatory diagnosis of hypothyroid conditions in pregnant women.
Footnotes
Acknowledgments
The authors are supported by the following grants: FONDECYT no. 11000926, FONDECYT no. 1070352, FONDECYT no. 1085281, FONDECYT no. 3070018, FONDECYT no. 3100090, FONDECYT no. 11075060, FONDECYT no. 3130539, Millennium Institute on Immunology and Immunotherapy (P-09-016-F), Millennium Nucleus (P07-011-F), and Proyecto Interno Universidad Andrés Bello DI-01-44-08. L.J.C. is a Pew Latin American Fellow in the Biomedical Sciences.
Author Disclosure Statement
No competing financial interests exist.