
Abstract
Background:
The atherogenic effects of hypothyroidism on lipid metabolism could result, in part, from the reduced clearance of remnant lipoproteins. In this study, we investigated the expression of hepatic low-density lipoprotein receptor–related protein 1 (LRP1), a receptor for remnant lipoproteins, in hypothyroidism and the effect of 3,3′,5-triiodo-L-thyronine (T3) treatment on hepatic LRP1 expression.
Methods:
C57BL/6 mice were fed a normal diet (control group) or a low-iodine diet supplemented with 0.15% propylthiouracil (PTU/LI group) for 4 weeks. Mice in the PTU/LI group were injected intraperitoneally with T3 (0, 30, and 150 μg/kg of body weight) for 7 days. HepG2 cells were incubated in fetal bovine serum or charcoal-stripped fetal bovine serum with various concentrations of T3. The expression and function of LRP1 in liver samples and cells were analyzed.
Results:
Hypothyroidism was successfully induced in PTU/LI mice. Hepatic LRP1 protein expression was lower in the PTU/LI group than in the control group. T3 treatment upregulated hepatic LRP1 protein expression in PTU/LI mice. LRP1 expression in HepG2 cells was reduced after incubation in the medium containing charcoal-stripped fetal bovine serum, which mimics hypothyroidism in vitro, and was recovered by T3 treatment. The protein expression of LRP1 in HepG2 cells was increased by T3 treatment in a dose-dependent manner up to 2.0 nM T3. However, LRP1 mRNA transcription was not affected by hypothyroidism conditions or T3 treatment, either in liver samples or in HepG2 cells. T3 treatment on HepG2 cells increased cellular uptake of lipid-conjugated apolipoprotein E through LRP1.
Conclusions:
Our data demonstrate that hepatic LRP1 expression and function decrease in hypothyroidism and are regulated by the thyroid hormone. These results suggest that in hypothyroidism, decreased expression of hepatic LRP1 may be associated with reduced clearance of circulating remnant lipoproteins.
Introduction
LDL receptor–related protein 1 (LRP1) is a member of the LDL receptor gene family. This cell surface glycoprotein is a multifunctional scavenger and signaling receptor that binds and internalizes diverse ligands (12). It is expressed on the surface of hepatocytes and binds lipid-conjugated apolipoprotein E (ApoE) and internalizes triglyceride-rich lipoproteins containing ApoE such as chylomicron remnants and very low-density lipoprotein remnants (12). The LDL receptor, which recognizes apolipoprotein B–containing lipoproteins such as LDL, also can bind lipidated ApoE, and both LRP1 and the LDL receptor in hepatocytes play important roles in the clearance of remnant lipoproteins (13 –17). Although LRP1 does not play a role in hepatic uptake of LDL and clearance of circulating LDL, this receptor is one of the major contributors to the clearance of remnant lipoproteins (accounting for ∼80% of the LDL receptor–mediated clearance of chylomicron remnants) (18).
As mentioned above, postprandial serum triglyceride levels are elevated in patients with hypothyroidism (3). This might be explained by decreased clearance of chylomicron remnants and VLDL remnants in hypothyroidism (2,4). Furthermore, thyroxine (T4) treatment has been shown to enhance clearance of these remnant lipoproteins in patients with hypothyroidism (2,4). Therefore, hepatic receptors contributing to the clearance of circulating chylomicron remnants could be altered in hypothyroidism. A number of studies have reported the upregulation of hepatic LDL receptors by the thyroid hormone and the specific regulatory mechanisms (19 –22). However, the effect of the thyroid hormone on hepatic LRP1 has never been investigated. We hypothesized that hepatic LRP1 expression and LRP1-mediated clearance of remnant lipoproteins would be reduced in hypothyroidism and that thyroid hormone replacement therapy would recover these alterations.
In this study, we investigated the effect of the thyroid hormone on the expression and function of hepatic LRP1 by performing in vivo and in vitro experiments. On the basis of our findings, we propose a novel mechanism for the development of atherogenic dyslipidemia in patients with hypothyroidism.
Materials and Methods
Cell culture and preparation
HepG2 cells were cultured in minimum essential medium containing 10% fetal bovine serum (Thermo Scientific, Rockford, IL) in 5% CO2/95% air at 37°C. 3,3′,5-Triiodo-L-thyronine (T3; Sigma-Aldrich, St. Louis, MO) was reconstituted with NaOH and prepared at −20°C. To mimic the hypothyroidism environment in vitro, HepG2 cells were incubated in 10% charcoal dextran–stripped fetal bovine serum (CSS; Gemini Bio-Products, Woodland, CA) for 24 hours. HepG2 cells incubated in CSS were incubated with the indicated concentrations of T3 for 48 hours by adding the stock solution to the culture medium. Phosphate buffered saline (Thermo Scientific) was used to dilute the T3 stock solution.
Animals, diet, and treatment
Laboratory animals were cared for in accordance with the National Institutes of Health's guidelines. The animals were maintained according to the ethics guidelines of our institution, and the experimental protocol was approved by the Committee on Animal Investigations of Seoul National University Bundang Hospital. Male C57BL/6 mice, aged 12–14 weeks, were housed in a temperature-controlled environment under a 12-hour light–dark cycle and allowed ad libitum access to diet and water. Seventeen mice were divided into two groups: a control group (normal diet, n=6; Purina irradiated laboratory chow 38057; Purina Korea, Seoul, South Korea) and a propylthiouracil/low-iodine (PTU/LI) group (n=11). The PTU/LI group mice were fed a low-iodine diet supplemented with 0.15% PTU for 4 weeks to induce hypothyroidism (23,24). The 11 mice in the PTU/LI group were divided again into 3 treatment groups according to the dose of T3 (0, 30, and 150 μg/kg). The physiological dose of T3 was calculated by normalizing the dose to the body surface area (25). T3 (Sigma-Aldrich) in 20% dimethyl sulfoxide was administered on a daily basis through intraperitoneal injection for 7 days, and all animals without T3 treatment were injected 20% dimethyl sulfoxide in the same way as T3-treated animals. All animals were sacrificed after physiological fasting for six hours (i.e., from 06:00 to 12:00). Livers were extracted, processed, and embedded in paraffin for histological analysis. The remaining tissues were flash-frozen in liquid nitrogen and stored at −80°C until analysis. Blood was collected by inferior vena cava puncture and stored at −20°C.
Serum lipid profiles
Blood samples were obtained at the time of sacrifice and were immediately centrifuged at 5000 g for 5 minutes. TC was measured using a Cholesterol Fluorometric Assay Kit (Cayman Chemical Company, Ann Arbor, MI) and triglycerides were determined using a Triglyceride Colorimetric Assay Kit (Cayman Chemical Company).
Immunohistochemistry
Mouse livers were paraformaldehyde fixed, paraffin embedded, and cut into 5–6 mm sections. The sections were stained with an antibody to LRP1 (1:100; Epitomics, Burlingame, CA). Staining was viewed using a 3,30-diaminobenzidine tetrahydrochloride kit (Evision plus kit; Dako, Glostrup, Denmark).
Total RNA and cDNA preparation
Total RNA was isolated from HepG2 cells and mice liver tissues using Trizol reagent (Invitrogen, Carlsbad, CA) and quantified using a NanoDrop® ND-1000 spectrophotometer (Thermo Scientific). After the RNA extraction, 4 μg of RNA was treated with 1 U RNase-free DNase I to remove all contaminating genomic DNA. After removing the DNase I, the DNase-treated RNA was subsequently used for cDNA synthesis using a moloney murine leukemia virus reverse transcriptase (Promega, Madison, WI) according to the manufacturer's protocol. The synthesized cDNA was stored at −20°C for later use.
Quantitative real-time polymerase chain reaction
Quantitative real-time polymerase chain reaction analysis was performed using a TaqMan assay kit for LRP1 (Hs00233856_m1, Mm00464608_m1) and an ABI 7500 instrument (Applied Biosystems, Foster City, CA). β-Actin (Hs99999903_m1, Mm00607939_s1) was used as an invariant control. Polymerase chain reactions were performed in triplicate in a final volume of 20 μL according to the manufacturer's protocol. For each assay, a standard curve was obtained by analyzing a series of dilutions of pooled cDNA samples for the relevant gene. Data were analyzed with Sequence Detector 1.7 software (Applied Biosystems). The results are expressed as the ratio of the expression of the gene of interest relative to the expression of β-actin.
Immunoblot analysis
Cell lysates were prepared using MPER® (Thermo Scientific), and aliquots of cell lysates and tissue homogenates were denatured under reducing conditions (1.75% sodium dodecyl sulfate, 15 mM 2-mercaptoethanol) for 5 min at 100°C. The total protein amount in each cell lysate was determined by the Bradford assay (Sigma-Aldrich). Cell lysates containing 10 μg of protein were loaded onto sodium dodecyl sulfate polyacrylamide gel electrophoresis gels for immunoblot analysis. To assess LRP1 expression, nitrocellulose membranes were incubated with an anti-LRP1 antibody (Epitomics) at a 1:1500 dilution overnight at 4°C, and then subsequently with horseradish peroxidase–conjugated goat anti-rabbit IgG (Santa Cruz Biotechnology, Santa Cruz, CA) at a 1:5000 dilution for 1 hour at room temperature. The signals were detected with the ECL Western Blotting Analysis System (Thermo Scientific). As a loading control, β-actin immunoreactivity was detected with a monoclonal anti-β-actin antibody (Sigma-Aldrich) at a 1:5000 dilution and horseradish peroxidase–conjugated goat anti-mouse IgG (Santa Cruz Biotechnology) at a 1:5000 dilution.
Small interfering RNA transfections
Small interfering RNA (siRNA) targeting human LRP1 (siLRP1), the LDL receptor (siLDLR), and nontargeting negative siRNA (siCTRL) were purchased from Thermo Scientific. Each siRNA was transfected into HepG2 cells using Lipofectamine 2000 (Invitrogen). We determined the siRNA silencing efficiency by the real-time polymerase chain reaction of target gene mRNA.
ApoE uptake analysis
HepG2 cells incubated in CSS were treated with the indicated concentrations of T3. If needed, siLRP1 or siLDLR was transfected into HepG2 cells to silence the LRP1 or LRL receptors before incubation in CSS and T3 treatment. After 48 hours of T3 treatment, cells were washed once with phosphate buffered saline, and then incubated with 25 μg/mL human recombinant ApoE3 (R&D Systems, Minneapolis, MN) for 1 hour. In these experiments, we used ApoE3 that was reconstituted with 1,2-dimyristoyl-sn-glycero-3-phosphocholine as described previously (26). Before incubation with lipidated ApoE, cells were incubated with 200 nM human recombinant receptor–associated protein (Merck Chemicals, Nottingham, United Kingdom) to block the LRP1 and LDL receptors for 30 minutes, if needed. For further analysis, HepG2 cells were washed three times with phosphate buffered saline and harvested with Mammalian Protein Extraction Reagent (Thermo Scientific). The lysates were subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis using an anti-human ApoE3 antibody at a 1:150,000 dilution (R&D Systems).
Statistical analysis
All statistical analyses were conducted using SPSS software (version 20.0; SPSS, Chicago, IL). Values are expressed as mean±standard error. Statistical comparisons between groups were performed using the Student's t-test. A p-value<0.05 was considered statistically significant.
Results
Hepatic LRP1 expression in a hypothyroidism animal model and the effects of thyroid hormone treatment on hepatic LRP1 expression in animals
We measured total T4 and free T4 levels in the serum of the control group mice and untreated mice in the PTU/LI group. Total T4 and free T4 levels were lower in the PTU/LI group than in the control group (total T4, control vs. PTU/LI, 1.53±0.43 vs. 0.97±0.21 μg/dL, p=0.034; free T4, 0.30±0.14 vs. 0.17±0.04 ng/dL, p=0.017), and hypothyroidism was successfully induced in the PTU/LI group mice. We also measured fasting serum TC and TG levels in the animals (Supplementary Fig. S1; Supplementary Data are available online at
Hepatic LRP1 protein expression was lower in the PTU/LI group mice than in the control group mice (Fig. 1A, B). However, LRP1 mRNA expression was not different between the PTU/LI group and the control group (Fig. 1C). The immunohistochemistry analysis also showed reduced hepatic LRP1 protein expression in the PTU/LI group (Fig. 2). We treated the PTU/LI group mice with 30 and 150 μg/kg of T3 for 7 days. Although hepatic LRP1 protein expression increased in the T3-treated mice compared with the vehicle-treated mice, there was no significant difference in LRP1 protein expression according to the dose of T3 treatment (Fig. 3A, B). T3 treatment did not change mRNA expression of hepatic LRP1 (Fig. 3C).
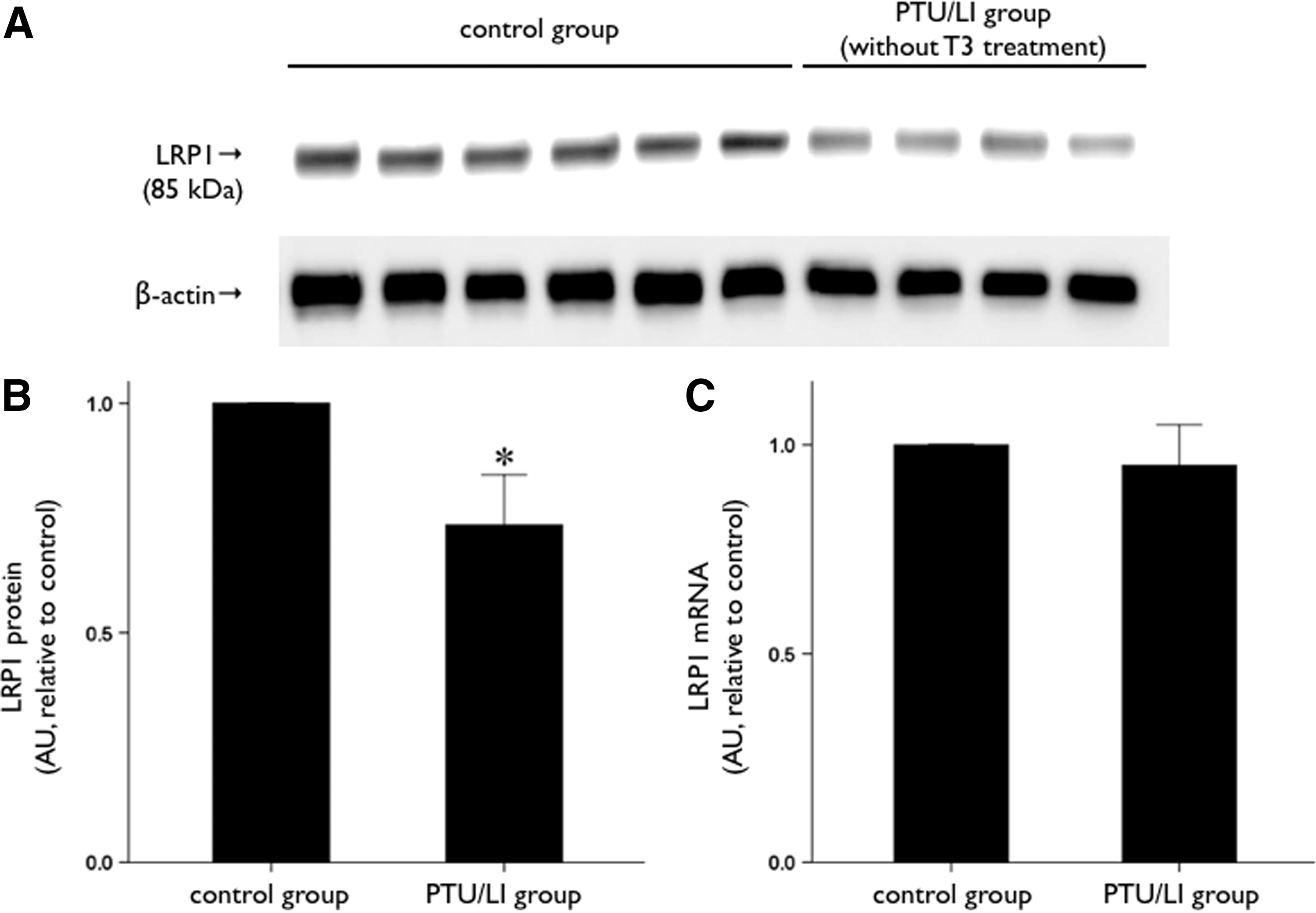
Change in hepatic LRP1 expression in a hypothyroidism animal model. C57BL/6 mice were fed a normal diet (control group, n=6) or a low-iodine diet supplemented with 0.15% propylthiouracil (PTU/LI group, n=4) for 4 weeks.
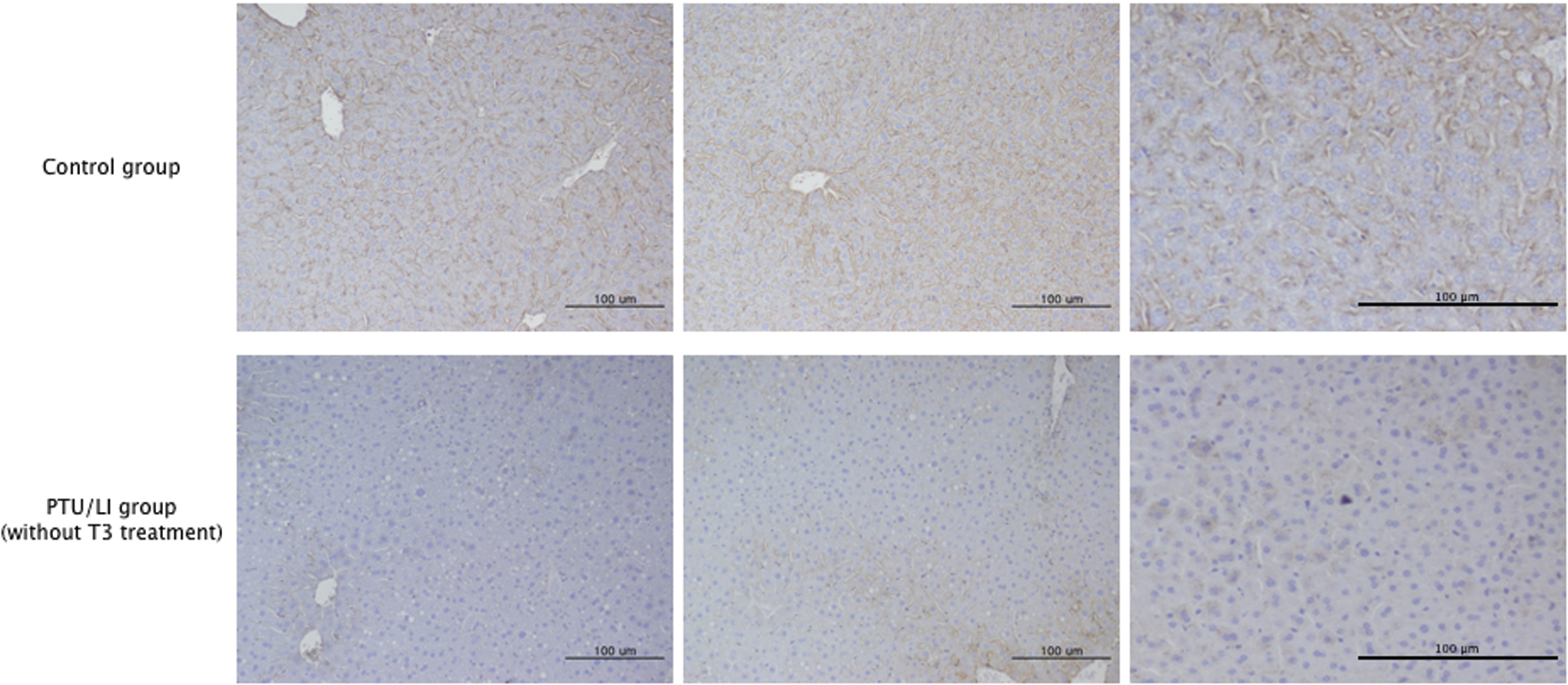
Immunohistochemistry of LRP1 in the liver samples. L C57BL/6 mice were fed a normal diet (control group, n=6) or a low-iodine diet supplemented with 0.15% propylthiouracil (PTU/LI group, n=4) for 4 weeks. Three samples of each group are presented and LRP1 expression is shown by brown color. Bars indicate 100 μm. Color images are available online at
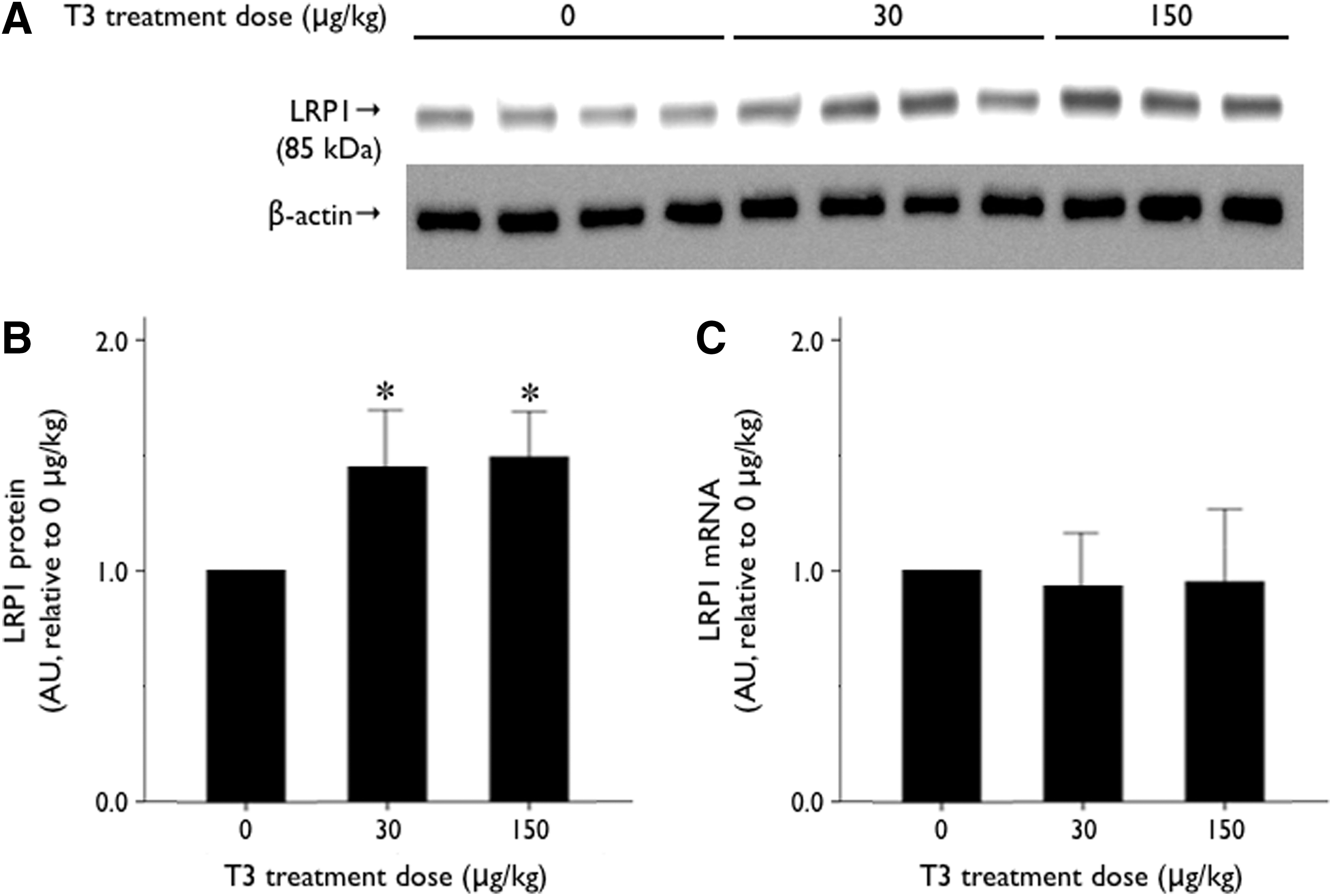
Change in hepatic LRP1 expression after T3 treatment in animals. C57BL/6 mice were fed a low-iodine diet supplemented with 0.15% propylthiouracil (n=11). Various doses of T3 (0, 30, and 150 μg/kg of body weight) were administered daily to the mice through intraperitoneal injection for 7 days.
The effect of the thyroid hormone on LRP1 protein expression in HepG2 cells
To mimic hypothyroidism in vitro, we used CSS. CSS contains no thyroid hormone because the thyroid hormone has been removed by charcoal. After 24-hour incubation in CSS, LRP1 protein expression was reduced in HepG2 cells (Fig. 4A). T3 treatment recovered the reduced protein expression of LRP1 in HepG2 cells (Fig. 4A). In addition, upregulation of LRP1 protein expression by T3 treatment was dose dependent up to 2.0 nM of T3 in the culture medium, and LRP1 protein expression plateaued at T3 concentrations greater than 2.0 nM in HepG2 cells (Fig. 4B). Treatment of HepG2 cells with various T3 concentrations did not change LRP1 mRNA expression (Fig. 4C).

The effect of T3 on LRP1 expression in HepG2 cells. HepG2 cells incubated in CSS or unstripped FBS for 24 hours were treated with the indicated concentrations of T3 for 48 hours.
The effect of the thyroid hormone on ApoE uptake through LRP1 in HepG2 cells
To investigate whether the thyroid hormone regulates not only LRP1 expression but also ApoE uptake by this receptor, we compared intracellular levels of ApoE in HepG2 cells incubated with various concentrations of T3. When we did not add lipid-conjugated ApoE to the culture medium, intracellular endogenous levels of ApoE were not increased by T3 treatment (Fig. 5A). However, intracellular ApoE was increased by T3 treatment in a dose-dependent manner when lipidated ApoE was added to the culture medium (Fig. 5A). These results demonstrate that T3 treatment upregulated ApoE uptake in HepG2 cells. We used siRNA targeting LRP1 to investigate whether this T3-induced increase of ApoE uptake was due to upregulation of LRP1 in HepG2 cells. As expected, T3-induced ApoE uptake was not observed in HepG2 cells when LRP1 was knocked down by siLRP1 (Fig. 5B). To exclude the effect of LDL receptor on ApoE uptake, we knocked down the LDL receptor in HepG2 cells using siLDLR. As shown in Figure 5C, ApoE uptake was increased by T3 treatment in HepG2 cells, and T3-induced ApoE uptake was inhibited by human recombinant receptor–associated protein, a functional blocker of the LRP1 and LDL receptors. These results were maintained even when the LDL receptor was knocked down in HepG2 cells.

Uptake of lipid-conjugated ApoE3 by T3 in HepG2 cells. HepG2 cells incubated in CSS for 24 hours were treated with the indicated concentrations of T3 for 48 hours. Human recombinant ApoE3 conjugated with lipid was added to the culture medium and cells were incubated for 1 hour.
Discussion
In this study, we demonstrate that LRP1 expression and function are regulated by the thyroid hormone. Despite the major role of LRP1 in the clearance of circulating atherogenic particles, few studies have investigated the regulation of LRP1. Llorente-Cortés et al. (27 –29) investigated the regulation of LRP1 in vascular smooth muscle cells and macrophages and reported that sterol regulatory element binding protein-2 negatively regulated LRP1 expression in macrophages (29), whereas in a previous study, we reported that sterol regulatory element binding protein-2 is a key activator of LRP1 expression in hepatocytes (30). In adipocytes, LRP1 is known to be regulated by the peroxisome proliferator–activated receptor-γ (31), and we also re-affirmed the regulation of LRP1 associated with peroxisome proliferator–activated receptor-γ in human brain microvascular endothelial cells (32) and hepatocytes (33). In the present study, we demonstrate that the thyroid hormone regulates LRP1 expression in hepatocytes. In a PTU/LI-induced hypothyroidism animal model, hepatic LRP1 protein expression was decreased compared with the control animals, and T3 treatment increased hepatic LRP1 protein expression in the hypothyroid animals. LRP1 protein expression in HepG2 cells incubated in the medium without the thyroid hormone was reduced compared with cells incubated in the control medium. T3 treatment also increased LRP1 protein expression in HepG2 cells. T3-induced LRP1 expression in HepG2 cells increased in a dose-dependent manner up to the T3 concentration of 2.0 nM and plateaued at T3 concentrations greater than 2.0 nM. Considering that the physiologic serum concentration of T3 is 1.0–3.0 nM in humans, our data suggest that a low T3 concentration in hypothyroidism can be associated with an alteration in hepatic LRP1 expression. T3 treatment of hepatocytes with thyrotoxic doses (above 10 nM of T3) was reported to increase oxidative stress and cellular damage; oxidative stress markers were maximally increased at 20 nM of T3, and 200 nM T3 produced 69% apoptosis after 24 hours of treatment (34). This thyrotoxic effect of T3 on hepatocyte might be one of the possible explanations for the plateau of the T3 effect on LRP1 expression over 20 nM observed in this study.
In our data, T3 treatment regulated LRP1 protein expression in hepatocytes, and the transcription of LRP1 mRNA was not affected by T3 treatment either in the liver samples from mice or in HepG2 cells. Classically, T3 regulates gene expression by binding to the high-affinity thyroid hormone receptors that recognize specific response elements in the promoters of T3-target genes (35). Hepatic LDL receptor expression is also regulated through this mechanism by the thyroid hormone (21,22). The thyroid hormone directly upregulates LDL receptor mRNA transcription (21), and indirectly increases hepatic LDL receptor mRNA transcription through the activation of sterol regulatory element binding protein-2 mRNA transcription (22). Although LRP1 is a member of the LDL receptor gene family, its regulation by the thyroid hormone does not appear to be mediated by direct or indirect genomic actions of the thyroid hormone on the LRP1 gene, because hepatic LRP1 mRNA transcription was not affected by T3 treatment. These data indicate that the thyroid hormone affects LRP1 translation or post-translational modifications in hepatocytes. In addition, the protein degradation of LRP1 could be regulated by the thyroid hormone. Although we demonstrated that hepatic LRP1 protein expression is regulated by the thyroid hormone, we were not able to elucidate the specific regulatory mechanism.
We also demonstrate that lipid-conjugated ApoE uptake via LRP1 is also regulated by the thyroid hormone in hepatocytes. Moreover, our data show that this regulation of ApoE uptake by the thyroid hormone is mediated specifically by LRP1. Both the LDL receptor and LRP1 are major contributors to the clearance of circulating remnant lipoproteins in the liver; these receptors contribute to 80% of hepatic uptake of circulating chylomicron remnants in mice (18). We found that T3 treatment increased both LDL receptor and LRP1 protein expression in HepG2 cells; the increase in LDL receptor expression by the thyroid hormone is consistent with the results reported in previous studies (21,22). The regulation of lipid-conjugated ApoE uptake by the thyroid hormone was mainly through LRP1 in HepG2 cells. ApoE uptake in HepG2 cells was increased by the thyroid hormone in a dose-dependent manner within the physiologic range of T3 concentrations. ApoE uptake was not altered by thyroid hormone treatment when LRP1 was knocked down by siRNA. In addition, expression of the LDL receptor did not affect the thyroid hormone–induced increase in ApoE uptake by HepG2 cells. These results suggest that LRP1, rather than the LDL receptor, may be the major contributor to the thyroid hormone–induced clearance of circulating remnant lipoproteins.
Our results have some important clinical implications. Hypothyroidism results in an atherogenic lipid profile by increasing fasting LDL cholesterol, apolipoprotein B, lipoprotein (a), and postprandial triglyceride levels (1 –4). The increase in postprandial triglyceride or triglyceride-rich remnant lipoproteins has been attributed to prolonged gastric emptying, increased intestinal absorption, increased production and excretion of chylomicrons, decreased lipoprotein lipase activity, or decreased hepatic clearance of triglycerides in the setting of hypothyroidism (3,9 –11). Our results suggest that alteration of hepatic LRP1 expression could be a novel mechanism resulting in the accumulation of circulating remnant lipoproteins in hypothyroidism. Considering that LRP1 plays a major role in increasing lipidated ApoE uptake after T3 treatment in HepG2 cells, regardless of the expression of the LDL receptor, the decrease in the hepatic clearance of remnant lipoproteins in hypothyroidism might be due mainly to a decrease in hepatic LRP1 expression. Recent studies have reported the association of hypothyroidism and nonalcoholic fatty liver disease (36) and the role of postprandial lipemia in the pathogenesis of nonalcoholic fatty liver disease (37). Our results of the alteration of hepatic LRP1 in hypothyroidism might present a clue for the molecular mechanism of the development of nonalcoholic fatty liver disease in hypothyroidism. In addition, LRP1 has diverse ligands and multiple functions in various tissues. LRP1 has been reported to be associated with the pathogenesis of Alzheimer's disease (38,39), vascular damage or atherosclerosis (40,41), and cancer cell migration and metastasis (42,43). The regulation of LRP1 expression by the thyroid hormone in hepatocytes may hold true for other tissues and cell types, which may explain the association between various medical conditions and thyroid dysfunction.
One of the limitations of this study is the lack of postprandial serum lipid profile data for the mice. Although we demonstrate the regulation of hepatic LRP1 protein expression by the thyroid hormone in vivo, we did not evaluate a possible effect of the thyroid hormone–induced changes in hepatic LRP1 expression on postprandial serum triglyceride levels or remnant-like particle-cholesterol levels. We only measured fasting serum TC and TG levels in this study, and it has been reported that fasting TG levels are not changed in hypothyroidism (44). Therefore, to confirm the causative effect of the thyroid hormone–associated hepatic LRP1 regulation on serum lipid profiles or cardiovascular disease risk, further studies are needed. Despite this limitation, the present study is meaningful because increased serum postprandial triglyceride and chylomicron remnant levels in hypothyroidism have already been reported in humans (2,3), and our in vitro data show that the thyroid hormone regulates lipid-conjugated ApoE uptake through LRP1 in HepG2 cells. Another limitation of this study is that we did not elucidate the specific regulatory mechanism of hepatic LRP1 protein expression by the thyroid hormone. As mentioned above, the thyroid hormone classically regulates gene expression by binding to the thyroid hormone receptors (35). Although some nongenomic mechanisms of the thyroid hormone effects have been suggested (35,45), further studies are also needed to elucidate how the thyroid hormone regulates LRP1.
In conclusion, our data demonstrate that hepatic LRP1 protein expression and LRP1-mediated cellular uptake of lipid-conjugated ApoE are regulated by the thyroid hormone. These results suggest that decreased hepatic LRP1 might be associated with the reduced clearance of circulating remnant lipoproteins, thereby altering the serum lipid profile in hypothyroidism.
Footnotes
Acknowledgment
This work was partly supported by a grant from Seoul National University Bundang Hospital for 2012 (Grant No. 11-2012-001).
Disclosure Statement
The authors have nothing to disclose.