
Abstract
Thyroid hormones (THs) are important in the development and maintenance of lipid and energy homeostasis. THs act through two closely related TH receptors (TRs α and β), which are conditional transcription factors. Recently, TH analogues or thyromimetics with varying degrees of TR subtype and liver uptake selectivity have been developed. These compounds exert beneficial effects of TH excess states without many undesirable TR-dependent side effects. Several selective TR modulators (STRMs) showed exceptionally promising results in lowering serum cholesterol in preclinical animal models and human clinical studies. Moreover, some first generation STRMs elicit other potentially beneficial effects on obesity, glucose metabolism, and nonalcoholic fatty liver disease (NAFLD). While it was initially thought that STRMs would be an effective long-term therapy to combat elevated cholesterol, possibly in conjunction with another cholesterol-lowering therapy, the statins, three major first generation STRMs failed to progress beyond early phase III human trials. The aim of this review is to discuss how STRMs work, their actions in preclinical animal models and human clinical trials, why they did not progress beyond clinical trials as cholesterol-lowering therapeutics, whether selective TR modulation continues to hold promise for dyslipidemias, and whether members of this drug class could be applied to the treatment of other aspects of metabolic syndrome and human genetic disease.
Introduction
Early attempts to use natural THs for prevention of heart disease and weight loss were not successful (12,13); harmful side effects outweighed beneficial effects (Fig. 1). The field of selective TH modulation nevertheless underwent a renaissance in the 1980s when novel findings suggested rational routes to create selective thyroid hormone receptor (TR) modulators (STRMs) with improved spectrums of actions. Development and actions of these compounds, their fates in preclinical and human clinical trials and possible clinical applications will be the subject of this review.
Timeline in the development of STRMs. Development of thyroid hormone receptor selective modulators through the last 50 years. TH, thyroid hormone; TR, thyroid hormone receptor; STRMs, selective thyroid receptor modulators.
Discussion
Thyroid hormone synthesis and activity
THs are secreted by the thyroid gland into the general circulation and this process is tightly regulated by a negative feedback loop (4,5,14). Synthesis and release of TH is triggered by interactions of pituitary thyrotropin (TSH) with a cognate receptor (TSHR) expressed on the surface of thyroid follicle cells. TSH synthesis and release is triggered by thyrotropin releasing hormone (TRH), secreted by the periventricular nucleus of the hypothalamus. Expression of the pituitary TSH and hypothalamic TRH genes is subject to negative transcriptional regulation by THs. This hypothalamic-pituitary-thyroid (H-P-T) axis maintains circulating TH levels within defined concentration ranges (referred to as “set point”).
Secreted THs exert pleiotropic actions on a variety of organs and cell types in the periphery and central nervous system. TH is secreted by the thyroid gland, predominantly in the form of thyroxine (T4). T4 is then converted into the main active form of the hormone (triiodothyronine [T3]) by deiodinases (DIO) 1 and 2 or converted into inactive forms through the actions of DIO3 (15). The effects of the three DIO enzymes are integrated to promote the maintenance of serum T3 concentrations in healthy subjects (16,17).
Availability of active T3 for nuclear TRs is regulated at several steps. Cellular TH uptake is mediated by specific membrane transporters, including monocarboxylate transporters (MCTs) 8 and 10 and organic anion transporter protein 1c1 (OATP1C1), which are differentially expressed and vary in substrate preferences (18). THs can be sequestered into inactive pools within the cytoplasm of certain tissues, including liver and kidney, via interactions with a NADP-regulated TH binding protein, also known as Mu-crystallin, and encoded by the CRYM gene (19). As already mentioned, T4 is converted to the active hormone T3 via actions of deiodinases, and this event can be rate limiting for activation of local TH signaling (16,17). Finally, T4 and T3 are subject to additional modifications, which mostly result in TH derivatives that display reduced binding affinity for TRs and reduced classical biological activity (16). One exception to this rule is that decarboxylation of T4 by aromatic amino acid decarboxylase coupled to DIO-mediated deiodination events results in 3-iodothyronamine and thyronamine, which regulate important aspects of body temperature and heart rate (20,21).
All of these steps are subject to additional regulatory inputs meaning that TH signaling can be selectively induced by controlling local T3 availability. For example, TH signaling is often selectively induced by signals that control DIO2 transcription (16,17). Bile acids act through a G-coupled protein receptor (TGR5) that is expressed on brown adipose tissue (BAT) to induce DIO2, thereby activating local TH signaling events required for mitochondrial uncoupling and heat production (22).
Thyroid hormone receptors
Physiologic effects of TH are mostly mediated by TRs, members of the nuclear hormone receptor superfamily (4,5). There are two TR genes, TRα and TRβ, with different patterns of expression in different tissues; for example, TRβ expression predominates in liver (23). TRα has one T3-binding splice product, TRα1, which is predominantly expressed in brain, heart, and skeletal muscle, and two non–T3-binding splice products, TRα2 and TRα3, with several additional truncated forms generated by differential splicing. TRβ has three major T3-binding splice products, the most widely expressed is TRβ1; TRβ2 is expressed primarily in the brain, retina, and inner ear; and TRβ3 is expressed in kidney, liver, and lung. TRβ1 and TRβ2 are expressed in the hypothalamus and pituitary and are key regulators of the negative feedback loop that affects the circulating levels of TH through the H-P-T axis.
The major ligand binding forms of TR act in similar manner (4,5). TRs bind specific DNA response elements and alter the transcription rates of nearby genes via recruitment of coregulator complexes, which influence local chromatin structure and modification state along with RNA polymerase II recruitment and processivity. Hormone binding changes the transcription rate by modifying receptor conformation to facilitate the exchange of coregulator complexes with different functions and, in some cases, by altering TR DNA binding activity.
There are strong similarities in structural organization of TRα and TRβ (24). Each adheres to the signature domain arrangement of the NR family with an N-terminal domain required for optimal transactivation, a central DNA binding function, and a C-terminal ligand-binding domain (LBD). Hormone-dependent conformational shifts in position of the C-terminal LBD α-helix (helix 12) are responsible for hormone-dependent exchange of coactivator proteins for corepressors (25).
There is also evidence for alternative TH signaling pathways that involve non-TR proteins (26); these are often distinguished from classical TR-mediated actions of THs on the basis of (i) rapid kinetics, alternative TH responses can be faster than typical transcriptional responses, and/or (ii) unusual TH ligand preferences, which do not resemble those of nuclear TRs. In general, however, mutational analyses and RNA interference studies have frequently revealed that TH-dependent effects upon metabolic response and differentiation require classic TRs (5).
Rationale for TRβ-selective ligand development
In the 1980s and 1990s, new developments in TR molecular biology and structure suggested new possibilities for selective modulation of TH signaling pathways. A major breakthrough was the cloning of TRα and TRβ and the realization that these two closely related TRs have different biological functions and tissue distributions (27). One of the first genomic studies on human patients with resistance to thyroid hormone revealed significantly elevated circulating TH levels and elevated heart rate, which were associated with mutations in TRβ (28). These findings suggested that (i) TRβ regulates feedback inhibition of TH synthesis through the H-P-T axis (2) and (ii) TRα mediates effects of TH excess upon heart rate (29). More recently, the introduction of mutations into TRα and TRβ in mice has allowed further understanding of TR isoform preferences of T3 actions and revealed a major role for TRβ in the regulation of serum cholesterol levels, the metabolic rate, and the suppression of the H-P-T axis (30 –35). This led to the suggestion that TRβ-selective ligands would reduce serum cholesterol without elevation of heart rate (36).
The two TR isoforms cooperate in different aspects of regulation of basal metabolic rate and heat production through BAT activation. For BAT activation, TRα is required for optimal adrenergic signaling (37,38) and TRβ directly induces transcription of mitochondrial uncoupling protein 1 (UCP1) (39), required for dissipation of potential energy of the mitochondrial proton gradient as heat (40). Although TRα plays a major role in the regulation of metabolic rate, administration of T3 to Trα knockout mice elicited increases in metabolic rate and body temperature (36,41). This led to the suggestion that TRβ-selective analogues might induce small, but tolerable increases in basal metabolic rate without significant effects on the heart (36,42).
Further genetic analysis of TR subtype function has revealed more detail about the TR-isoform selectivity of TH actions in other tissues including regions of the brain, sensory organs, intestine, lung, cartilage, and bone, and this has been reviewed in detail elsewhere (5). One example is that TRα predominates in bone and plays a major role in skeletal development (43,44). Phenotypes of Trα knockout mice or mice that express dominant negative TRα resemble those of hypothyroidism and suggest that TRα is required for bone ossification, the process by which mesenchymal stem cells develop into cartilage that subsequently becomes mineralized. Further, impaired TRα activity results in reduced bone resorption and elevated bone mass in adults. Conversely, Trβ knockout mice display developmental bone phenotypes that include short stature, advanced bone age and premature ossification, and increased mineralization due to accelerated chondrocyte differentiation and increased osteoclast numbers (43). Adult Trβ knockout mice display increased bone resorption, opposite to phenotypes of adult Trα knockout mice. While TRβ is expressed in different compartments and cell types of the bone, these bone phenotypes resemble those of hyperthyroidism and probably reflect indirect consequences of disruption of TRβ-dependent regulation of the H-P-T axis, elevated circulating TH and hyperactivation of TRα.
Recent descriptions of inherited inactivating human TRα mutations have underscored the notion that the TRs play different roles in humans (45,46). These patients exhibit short stature and delayed bone development along with delayed cognitive development, confirming the importance of TRα in bone and neural development. Circulating TH levels are normal, consistent with the predominant role of TRβ in feedback inhibition of TH synthesis.
STRM development and first generation TRβ-selective STRMs
STRM development initially focused upon identification of TRβ-selective ligands. Structural analysis of TRα and TRβ LBDs suggested that this might be difficult; TR LBDs are highly homologous, with 32 single amino acid substitutions from a total of 237 amino acids (24). Further, X-ray structural analysis of TR LBDs revealed that only one of these subtype-specific amino acids (Ser277α/Asn331β) comprises part of the TRβ ligand-binding pocket defined in the presence of T3 or similar ligands.
Some selective TR ligands were created before fully rational design approaches [reviewed by Webb (47)]. The first TH analogues that lower cholesterol with reduced effects on heart were developed in the mid-1980s (48 –51). Prior to TR cloning of TRs and the realization that there are two TRs with different tissue distributions, L-94901 was reported to bind strongly to hepatic TRs but weakly to cardiac TRs (48). A Ciba-Geigy compound, CGS 23425, was confirmed as weakly TRβ selective (49). Both compounds lowered serum cholesterol without detrimental effects on the heart, and in addition, CGS 23425 was also confirmed to lower total serum and low-density lipoprotein (LDL) cholesterol and increase synthesis of apolipoprotein A1 (APOA1), the major protein component of high-density lipoproteins (HDL). Rare natural forms of active TH, such as Triac (3,5,3′-triiodothyroacetic acid), were also shown to be weakly TRβ selective and could lower cholesterol with reduced effects on heart rate (50).
Early synthetic ligands were discontinued for undisclosed reasons, but their actions confirmed that selective TR modulation is possible. Newer efforts at ligand design set out to introduce features of earlier compounds into new scaffolds and to further elaborate on these compounds. These efforts led to generation of several designed TRβ-selective ligands (Table 1), including the synthetically amenable scaffold GC-1 (5- to 10-fold selective) (52), the GC-1 derivative, GC-24 (40-100 fold selective) (53), KB141 (14 fold selective) (54) and KB2115 (TRβ selectivity not disclosed) (55).
LDL, low-density lipoprotein; LPA, apolipoprotein(a); TGs, triglycerides; NAFLD, nonalcoholic fatty liver disease; FH, familial hypercholesterolemia; ALD, adrenoleukodystrophy; DITPA, 3,5-diiodothyropropionic acid; T4, thyroxine; T3, triiodothyronine.
Molecular mechanisms of TRβ selectivity
X-ray crystallography provided insights into different modes of TRβ-selective binding (56). Binding modes of GC-1 and KB141 adhere to a classic “lock and key” model and both ligands display better fit in the TRβ ligand-binding pocket versus that of TRα (52,54). Here, indirect influences of the TRβ-specific pocket residue Asn331β induce favorable positions of nearby conserved arginine residues allowing for optimal interactions with the negatively charged GC-1 carboxylate group (24,57). Chemical modification of GC-1 confirmed that this substituent is required for optimal TRβ selectivity (58). GC-24 exploits a different and highly unexpected mode of selectivity; it contains a large phenyl extension group on the outermost thyronine ring, which exploits contacts with a TRβ-selective extension to the classical ligand binding pocket that only forms in the presence of this compound (53). It is thought that TRβ-selectivity arises because the equivalent region of TRα, comprised of exactly homologous amino acids, is more stable than that of TRβ, and this is probably a consequence of indirect distant influences of TR subtype-specific amino acids (59,60). Finally, Triac has an unique mode of selectivity, even though it resembles the agonist GC-1 and contains a negatively charged carboxylate group that is required for TRβ-selective binding (61). Here, X-ray structures reveal that there is TRβ-selective expansion of the pocket near the Triac carboxylate group and this effect permits large numbers of water molecules to enter the ligand-binding cavity. Ligand–water contacts compensate for lack of direct ligand–receptor contacts and also allow for increased mobility of the Triac carboxylate group in the TRβ pocket relative to that of TRα, the entropic contributions of the latter effect appear to explain TRβ subtype-selective ligand binding (61).
TRα-selective modulators and antagonists
In addition to TRβ-selective STRMs, synthetic chemistry efforts resulted in ligands with alternate modes of action. Scanlan and colleagues produced CO-23, which displays TRα-selective actions in cultured cells and frogs (62,63). CO-23 does not preferentially bind to TRα, indicating that subtype selectivity is a consequence of differential effects upon unknown steps involved in receptor activation rather than improved TRα subtype affinity. The Scanlan group has also generated TR antagonists (64,65) based on the so-called extension hypothesis, which states that ligands that resemble agonists but contain chemical extensions designed to dislodge LBD C-terminal helix 12 will induce transcriptionally inactive receptor conformations (66). The lead antagonist, NH-3, binds TRs with submicromolar affinity and blocks T3 response in frogs and rodents (67,64).
Both classes of compounds remain at the early developmental stage, but improved versions could be useful for certain clinical indications (66,68). TRα-selective agonists could be targeted to particular tissues to increase fat burning and metabolic rate without increasing heart rate and could also form scaffolds for synthesis of TRα-selective antagonists, which could be used to combat cardiac arrhythmias, even in euthyroid conditions. Existing TR antagonists, which are weakly TRβ selective, could be used as rapid-acting treatments for hyperthyroidism; current approaches are slow acting because of the long half-life of T4 in the circulation. TR antagonists could also be used to block tissue damage at sites of injury by slowing local metabolism and oxygen consumption.
Tissue-selective STRMs
Another way to obtain selective TR modulation of serum lipids is to direct TH analogues to tissues of interest, especially the liver, and away from tissues in which they induce harmful side effects. This approach was pioneered in the 1980s by Ciba-Geigy, which created CGH-509A, a conjugation product of L-T3 with cholic acid to promote liver uptake (2,51). This compound elicited significant reductions in serum cholesterol without effects on heart rate but was discontinued. More recently, a group at Metabasis also set out to generate TR agonists that specifically target the liver. These efforts yielded a so-called HepDirect prodrug MB07811, which undergoes first-pass hepatic extraction and cleavage by cytochrome P450, to generate the free methylphosphonic acid, MB07344, an active thyromimetic (69). MB07344 is eliminated in bile to escape the enterohepatic recirculation. A major amino acid transporter responsible for cellular uptake of T3 (monocarboxylate transporter-8) is unable to recognize and efficiently transport MB07344, ensuring that liver import is dependent upon HepDirect targeting (69).
Overlaps between modes of STRM selectivity and differences between STRMs
Interestingly, modern STRMs (such as GC-1, KB2115, and MB07344) display unexpected overlaps in their modes of specificity that were not intended design features. Analysis of tissue distributions of TRβ-selective STRMs (GC-1, KB2115) revealed that they display preferential uptake in liver; in fact, KB2115 is described as a highly liver-selective TH analogue (2). Of TRβ-selective ligands, only KB141 exhibits a wider tissue distribution pattern that resembled that of T3, as determined by mass spectroscopic analysis of tissue extracts (36). Mechanisms of liver-selective uptake of GC-1 and other TRβ-selective STRMs are unknown, but may be related to combinations of liver first-pass uptake and differential interactions with TH transporters. MB07344, the active product of the liver targeted prodrug MB07811, is more than 10-fold TRβ selective (70). Thus, first-generation modern STRMs combine different degrees of TRβ and liver uptake selectivity.
Other differential STRM properties should be considered when interpreting their actions, relative to each other and to native THs. STRMs bind to TRβ with different affinity from native hormones. While GC-1 binds TRβ with similar affinity as T3 (52), other STRMs display lower affinity [GC-24 (53); 2xT3, MB07344 (69,70); 2xT3 and KB141 (54), 6xT3]. Likewise, pharmacokinetic and pharmacodynamic properties of STRMs, not usually found in the public domain with the notable exception of MB07344 (69), could influence apparent ligand activities. Finally, STRMs can suppress TH production via effects on the H-P-T axis, hence it is important to confirm that STRM effects are consequences of direct TR actions and not indirect effects of hypothyroidism.
While TRβ-selective STRMs are often used as tools to probe mechanisms of T3 effects, great caution is required in drawing conclusions from the data. For example, as discussed in more detail below, GC-1 lowers cholesterol without harmful effects on heart rate (2). We currently favor the idea that liver-specific targeting is the key for safe cholesterol reduction, but it is difficult to understand whether this differential effect is a consequence of modest TRβ selectivity, liver uptake selectivity, or both. It is also hard to interpret studies that compare equimolar amounts of T3 and particular STRMs in animal models. Here, lack of an observed T3-like effect with a TRβ-selective STRM could mean that T3 actions are truly independent of TRβ or that differential tissue penetration, receptor affinity, pharmacodynamics, and pharmacokinetics means that equivalent levels of TR occupancy were not achieved in a given tissue in the experimental model.
Previous studies suggested that STRMs may not always behave as pure T3-like agonists at all gene loci [reviewed by Baxter and Webb (2)]. Our genome-wide comparisons of actions of saturating levels of T3 and GC-1 revealed that such effects were subtle and rare; for example, GC-1 exhibits reduced potency relative to T3 at the ANGPTL4 gene but not other genes (71). Emerging evidence that the two TRs display different activities at some genes could mean that more STRM-specific effects would emerge at doses that selectively activate TRβ versus TRα (71 –73), but this has not been proven.
While STRMs are a useful tool to probe TR actions in vivo, conclusions about TR subtype selectivity must be coupled with extensive consideration of phenotypes of Tr knockout mice and the direct analysis of STRM effects in vitro.
Actions of TRβ/liver-selective STRMs on lipid metabolism in animals
It was originally envisaged that STRMs would be long-term therapeutics for dyslipidemias, analogous to another widely used class of drugs, the HMG-CoA reductase inhibitors, or statins (2). Early short-term studies in preclinical animal models were successful (2,74). STRMs reduced total serum cholesterol in rodents (69,75 –77). Rodents are not ideal models to test cholesterol-lowering therapeutics, however, because most serum cholesterol is bound within the HDL fraction. Nevertheless, STRMs also elicited reductions in LDL and very low density lipoprotein (VLDL) fractions, and these effects became even more apparent after altering feeding regimens to induce hypercholesterolemia. Additionally, there were beneficial effects upon serum triglycerides (TGs) and nonesterified fatty acids in rodents and similar STRM effects were also documented in hamsters and rabbits (78). GC-1, KB141, and MB07811 reduced serum cholesterol in primates, where cholesterol fractions display a more “human-like” profile with higher proportions in the LDL fraction. STRMs also elicited beneficial reductions in levels of serum lipoprotein (a) [Lp(a)], an LDL-like particle confined to humans and primates that is an independent risk factor for atherosclerosis (2). Effects of STRMs were rapid, and changes were observed within 1–2 weeks and correlated with evidence for reduced atherosclerosis after extended treatments (78).
Dissection of mechanisms of TR cholesterol-lowering effects is far from complete but already suggests that STRMs regulate different steps of cholesterol metabolism from statins (Fig. 2). Verified T3-regulated genes that could influence cholesterol metabolic pathways include the LDL receptor gene (LDL-R), which promotes cholesterol reabsorption, and CYP7A1, which catalyzes the main step in cholesterol to bile acid conversion (79). Conversely, statins suppress liver HMG-CoA reductase activity (80), reduce liver cholesterol, and induce compensatory increases in liver LDL-receptor expression. This led to predictions that STRMs might either work additively or synergistically with statins, and this prediction was confirmed in rodents and in primates with MB07344 (81).
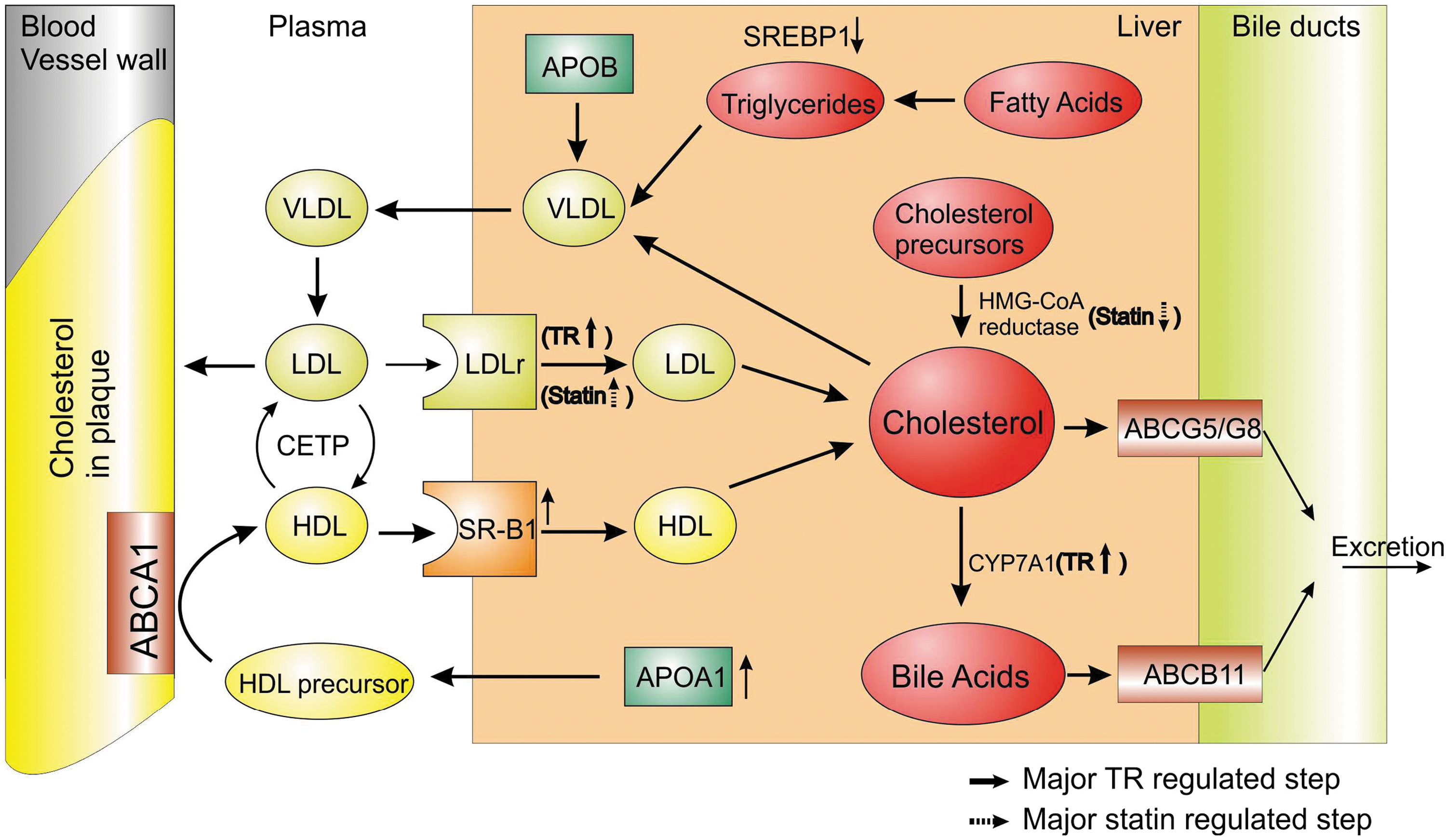
Schematic representation of actions of thyroid hormone (TH)/thyromimetics and statin on cholesterol metabolism. TH/thyromimetics and statins regulate different pathways in the cholesterol metabolism. STRMs and statins increase the hepatic expression of LDL receptor (LDLr) resulting in the plasma clearance of LDL cholesterol. STRMs also increase the hepatic expression of scavenger receptor type B1 (SR-B1), which promotes taking up of high-density lipoprotein (HDL) cholesterol particles brought back from the peripheral tissues like the atherosclerotic plaque to be taken up again in to the liver through a mechanism of reverse cholesterol transport. TH/thyromimetics also induce hepatic cholesterol 7 alpha-hydroxylase (CYP7A1), the rate-limiting enzyme in the conversion of cholesterol into bile acids. ATP-binding cassette transporter B11 (ABCB11) is a major determinant of bile flow, while transporters ABCG5/G8 are involved in the disposal of cholesterol directly as neutral sterols into the bile. The enzyme 3-hydroxy-3-methyl-glutaryl-CoA (HMG-CoA) is the rate-limiting step in the cholesterol synthesis and is inhibited by the statins, the current pharmaceutical drug for lipidemias. Other transporters shown are ATP-binding cassette transporter A1 (ABCA1) and cholesterol ester transfer protein (CETP). Thin arrows indicate steps known to be influenced by thyroid hormone mimetics, and up-regulated and down-regulated steps are distinguished by the direction of the arrow. Steps known to be influenced by statins are represented by dashed arrows. LDL, low-density lipoprotein; VLDL, very low-density lipoprotein.
Our laboratory demonstrated that GC-1 elicited massive cholesterol reductions in an LDL-R deficient mouse model fed a cholesterolemic diet (82), and similar effects were seen in Ldl-R knockout mice after high-dose administration of THs (83). These effects were accompanied by increases in markers of bile acid synthesis and excretion. Thus, the LDL receptor is not absolutely required for STRM- and T3-dependent cholesterol reduction, and induction of CYP7A1 probably plays a prominent role in cholesterol-lowering effects of STRMs in rodents.
There is also evidence that STRMs increase HDL turnover. TRs regulate synthesis of ApoA1 (84) and reabsorption of HDL into the liver (84). While it is not proven that STRMs induce reverse cholesterol transport, which removes cholesterol from atherosclerotic plaque and is a major target for pharmaceutical development, such effects would constitute important steps in up-regulating this process (85).
Beneficial effects of STRMs on serum lipids can be separated from harmful T3-like effects
STRM effects on serum lipids have consistently been obtained at doses that did not elicit the most undesirable TH-like effects on heart rate and muscle and bone catabolism in animal models. Several STRMs reduce cholesterol without detectable effects on heart rate in vivo or ex vivo (36,54,69,86,87). Unlike T3, analysis of blood chemistries after STRM treatment reveals no evidence for enhanced muscle catabolism. Moreover, GC-1 spares lean muscle mass during extended treatments in rodents (88), and GC-24 does not significantly alter skeletal muscle composition, as judged by fiber-type analysis of soleus and extensor digitorum muscles (89). Both ligands also lack T3-like effects upon muscle gene expression. GC-1 also lacked thyrotoxic effects upon bone mass as judged by dual-energy X-ray absorptiometry of several skeletal sites and histomorphometry of the distal femur (90). Overall, GC-1 and KB141 exhibited a 30-fold therapeutic window between cholesterol reduction and effects on heart rate in rodents and larger safety windows in primates, meaning that it is relatively easy to find doses of ligands that are sufficient to elicit lowering of cholesterol without a high risk of adverse effects (2). The highly liver-selective prodrug MB07344 exhibited even larger safety windows between cholesterol-lowering effects and harmful side effects in rodents (70); no effects on muscle and bone were observed at therapeutic doses.
Most STRMs suppress the H-P-T axis and lower serum T4/T3 levels to some extent, especially at high doses (2). Effects of the HepDirect prodrug MB07811 were less marked than other STRMs, probably because it is specifically targeted to liver (69). MB07811 did reduce serum T4 levels, and this effect was attributed to induction of DIO1 in liver and enhanced T4 to T3 conversion.
It is hard to predict consequences of long-term H-P-T axis suppression by STRMs. Since STRMs exhibit unique tissue distributions, extended STRM treatment should result in a mix of hyper- and hypothyroid effects in different tissues. Physiologic outcomes will therefore be specific to the STRM and the duration of ligand treatment.
Beneficial effects of TRβ/liver-selective STRMs upon metabolic syndrome
TH excess increases basal metabolic rate, oxygen consumption, and heat production, which lead to an increase in fat burning (2). The mechanisms appear pleiotropic, but as mentioned above, indications from Tr knockout mice suggest that TRα and TRβ are both involved in TH-dependent regulation of energy balance. This led to the suggestion that TRβ-selective analogues might induce weight loss without detrimental effects on the heart (91). Both GC-1 and KB141 dramatically reduce body fat in rodent models of obesity (Ob/Ob mice, Zucker rats, mice with diet induced obesity), lean rats, and primates without effects on lean muscle or bone (76,88). For GC-1, effective doses required for body fat reductions appear higher than those required for cholesterol reduction, but there is nevertheless a significant safety range between weight loss and cardiac effects (2).
It is likely that weight loss requires extrahepatic actions of STRMs. MB07811 (TRβ and highly liver selective) did not elicit significant weight loss in rodents, whereas KB141, which is predominantly TRβ selective and nontissue selective, did induce weight loss and increases in metabolic rate in comparative assays (69). Tissue targets for weight loss are not absolutely clear. GC-1 and GC-24 do not appear active in muscle, which is usually responsible for a large proportion of energy consumption. Another possibility is that STRMs activate BAT, which mediates heat production via uncoupled respiration in a process that is regulated by T3 and adrenergic signaling pathways. Indeed, GC-1 and GC-24 activate BAT and induce UCP1 in rodent models (88,91). Beiging of white adipose tissue (WAT) also contributes to elevated heat production and energy consumption (92). In this process, WAT acquires certain characteristics of BAT, including increased mitochondrial numbers, elevated UCP1 expression, and mitochondrial uncoupling. Since extended TH treatment increases mitochondrial numbers in adipose tissue, it will be interesting to investigate effects of STRMs upon beiging of WAT (93).
Whereas TH excess is usually associated with impaired glucose tolerance, due to impaired insulin action and enhanced gluconeogenesis in liver (94), three ligands that enhance BMR (GC-1, KB141, and GC-24) elicit substantial reductions in blood glucose levels in rodents. Given that MB07344 did not share these effects, and that hepatic actions of STRMs should enhance expression of gluconeogenic genes, we suspect that blood glucose-lowering actions are extrahepatic in origin. One obvious possibility is that blood glucose lowering is secondary to weight loss. Since increased thermogenesis may also result in insulin-independent lowering of blood glucose (95), BAT activation or adipose tissue beiging may be important for glucose lowering.
STRMs also exert beneficial effects on another aspect of metabolic syndrome, and this appears due to direct hepatic actions. Nonalcoholic fatty liver disease (NAFLD) is a major risk factor for obesity-associated cirrhosis. GC-1 prevented and reversed NAFLD in rats fed on diet deficient in choline and methionine and also decreased liver fat in cholesterol fed mice and mice with diet induced obesity (96). MB07811 (97) also exhibited strong antisteatotic activity in several normal and metabolically challenged animal models, confirming direct hepatic effects. In fact, MB07811 was even more potent than equivalent doses of T3, presumably because of its enhanced liver uptake. Several mechanisms have been proposed to explain prevention and reversal of NAFLD, and these include increased hepatic mitochondrial respiration rates and increased lipid oxidation. It may also be important to interpret actions of STRMs in the light of the recent discovery that TR activation clears liver fat by activating local autophagic pathways (98). NAFLD is an important cause of insulin resistance and type 2 diabetes (99); it is also possible that STRM-dependent improvements in insulin resistance could reflect tissue-selective effects on liver fat accumulation.
First-generation STRMs in human clinical trials
Three aforementioned designed STRMs (KB2115, GC-1, and MB07811) reached human clinical trials for dyslipidemias and displayed impressive early results for this indication (2,55,100). In these trials, which lasted up to 3 months, beneficial effects on serum lipids were observed in the absence of harmful effects that are typically associated with TH excess states.
KB2115 (KaroBio; Eprotirome) advanced into a phase III clinical trial (55,100). In initial clinical studies with KB2115, overweight male subjects received low doses (up to 200 μg/d) for 2 weeks, and these promoted lowering of LDL (55). The investigators did not observe effects on HDL levels, heart rate, or suppression of the H-P-T axis. A 12-week, placebo-controlled, double blind, randomized clinical phase II study was performed in 99 patients with high cholesterol levels to explore whether a clinically relevant LDL cholesterol–lowering effect can be achieved without affecting the heart, bone, muscle, and thyroid function. A pronounced and clinically relevant lowering of LDL cholesterol was observed, while the thyroid homeostasis was preserved outside the liver and biomarkers for heart, bone, muscle, and other organs were kept at normal levels.
In a phase IIb clinical study, 189 patients were treated with up to 100 μg KB2115 per day for 12 weeks in combination with a statin (100). This revealed that LDL cholesterol reductions could be obtained even alongside statins. Additionally, this study detected reductions in TGs and Lp(a). There was some suppression of T4 levels, but not of other markers of H-P-T axis activity, and no obvious elevation of heart rate or serum metabolic markers of bone catabolism.
In 2011, a 12-week placebo-controlled, double-blind, randomized, parallel-group, long-term phase III trial was initiated to assess the safety and efficacy of eprotirome in patients with heterozygous familial hypercholesterolemia who were receiving an optimal standard of care. The primary endpoint was the percent reduction in LDL cholesterol from baseline to 12 weeks. The development of eprotirome was discontinued in February 2012, after a 12 month dog toxicology study revealed that the exposure to the drug led to cartilage damage. This led to termination of this trial and all other related studies.
Phase I trials with GC-1 yielded results similar to KB2115 (2). Two phase I human trials were performed with GC-1 (QuatRx; sobetirome). Lipid-lowering effects in single and multiple doses were observed in the randomized, double-blind, placebo-controlled phase I studies of sobetirome. In the single dose study of 32 subjects, LDL cholesterol levels were decreased up to 22%, compared to 2% in subjects who received placebo. In the two week multiple dose study of 24 subjects, reductions in LDL cholesterol levels decreased up to 41% at doses up to 100 μg once a day, compared to an overall 5% reduction for placebo. Sobetirome was generally well tolerated and no cardiac abnormalities were reported (2, 101).
In 2006, a phase Ia clinical trial demonstrated that MB07811 was safe and well tolerated when administered as a single dose. In a 14 day phase Ib clinical trial, in which 56 subjects received multiple-dose clinical trial in subjects with mild hypercholesterolemia, MB07811 was also shown to be safe and well tolerated across all doses tested, ranging from 0.25 mg up to 40 mg. The frequency of other adverse events in MB07811-treated subjects was similar to placebo-treated subjects. No differences in heart rate, heart rhythm or blood pressure were observed between MB07811 and placebo-treated patients. The results of this clinical trial provide evidence that MB07811 lowers LDL cholesterol and TG levels in patients with mild hypercholesterolemia without affecting heart rate, heart rhythm or blood pressure.
Thus, all three STRMs reduce serum LDL cholesterol levels and other serum lipids without harmful TH-like effects in short term clinical trials. Very similar degrees of LDL cholesterol lowering (∼40%) are claimed with KB2115, GC-1 and MB07344 in human subjects (2). This is highly suggestive of a common mechanism of action.
DITPA: an STRM with a different profile
Results of human studies with KB2115 and other STRMs can be compared and contrasted with another TH analogue, DITPA (3,5-diiodothyropropionic acid) (100,102). DITPA is a low-affinity TR agonist with slight preference for TRβ versus TRα and no reported tissue uptake preference. DITPA was originally proposed as a therapy for heart failure; it had been shown to improve left ventricular function and increase cardiac output in animal models. While improvements in cardiac endpoints were not reproduced in humans, DITPA did lower circulating total and LDL cholesterol concentrations by 30% after a 24-week therapy, displayed similar effects in patients receiving statins, and lowered serum TGs (102). This profile resembles KB2115, implying that lipid-lowering actions and additive actions with statins are likely to be a feature of many TH analogues in humans. However, actions of DITPA differed from KB2115 in several ways (103). There was significant weight loss, though this study did not define which tissues accounted for reduction in body mass. DITPA also suppressed the H-P-T axis more strongly than KB2115. There were also increases in metabolic markers of bone degradation, a major counter-indication against long-term use of DITPA in humans. Finally, DITPA was very poorly tolerated. At doses used, only 37% of initially enrolled patients remained on therapy for the full 24 weeks. It is not clear why. One possibility is that weight loss was associated with elevated temperature. However, other adverse events, including gastrointestinal complaints, fatigue, and falls were more frequent in the DITPA group, suggesting that DITPA may induce a range of effects that led patients to drop out of the trial.
As already discussed, comparisons of different STRMs and thyromimetics may be difficult without knowledge of actual occupancy of TRs in target tissues, but our interpretation of DITPA and KB2115 exhibiting different profiles in humans is that liver selectivity of KB2115 mitigates against extrahepatic effects upon weight loss, H-P-T axis suppression, bone catabolism, and overall tolerability.
Fates of thyromimetics
Despite the promise of thyromimetics, it is not likely that any first-generation synthetic ligands that reached human clinical trials will develop into therapeutics. As mentioned, KB2115 was discontinued after long-term (12-month) dosing in dogs revealed adverse effects on cartilage at all doses. No phase II trials for GC-1 are currently planned for dyslipidemias. Most likely, this is due to strict regulation by the FDA of the approval of thyromimetics as long-term therapies, based on previously reported adverse cardiac side effects, which has made progress from concept to clinic expensive and long. Planned phase II clinical trials for MB07344 were terminated prior to initiation as the developing company Metabasis Therapeutics Inc. was acquired by Ligand Pharmaceuticals Inc. and further clinical trials were not continued.
The abrupt termination of the KB2115 phase III trial was a major setback for the thyromimetic drug class and it is important to understand adverse effects on cartilage. Presently, details of the dog trial have not been reported. It is not clear whether this adverse effect is a consequence of KB2115 interactions with TRs or adventitious interactions with another protein, whether it is drug or drug class specific, or whether the effect would be observed in humans. Our current attempts to understand this effect must therefore be restricted to speculation.
It is hard to understand why a highly liver-selective agonist with no obvious effects on bone catabolism would adversely influence cartilage. There are several hypotheses. As already mentioned, TRα plays an important role in cartilage formation in the context of bone ossification. Thus, one possibility is that KB2115 could bypass the liver to act directly on cartilage through TRs, either to superactivate normal TH signaling pathways or to reverse important actions of unliganded TRs. In this regard, KB2115 could act upon mature cartilage or upon mesenchymal stem cell precursors. KB2115 could also influence cartilage indirectly by inducing mild local or systemic hypothyroidism; as mentioned, there was weak suppression of T4 levels during human clinical trials with KB2115 (100), although this was not associated with changes in serum T3. Finally, and perhaps most speculatively, KB2115 could act in the liver to influence cartilage. Changes in the T4/T3 ratio during the aforementioned human clinical trial were attributed to a combination of weak suppression of the H-P-T axis and KB2115-dependent enhancement of DIO1 expression with concomitant enhanced T4 to T3 conversion in liver. Perhaps inappropriately elevated T3/T4 ratios could induce hyperthyroid effects on sensitive tissues, including cartilage. Alternatively, liver-selective actions of KB2115 could alter production of secreted liver proteins or other factors that influence cartilage.
Generally, the ways in which changes in TH signaling influence cartilage are poorly understood. It is clear that changes in TH balance alter processes involved in bone maintenance and formation (43,44). Moreover, emerging evidence suggests that DIO2 and DIO3 are susceptibility loci for osteoarthritis and that T3 availability regulates the biology of osteoblasts and chondrocytes and processes involved in maintenance and repair of bone in humans (104). The apparent defect that emerged in dogs after prolonged KB2115 treatment was, however, in connective tissue cartilage. Although some studies linked overt or subclinical hypothyroidism to cartilage defects (105), much less is known about TH effects on formation and maintenance of connective tissue cartilage versus bone development. The key to understanding why KB2115 affects cartilage is to define the precise cartilage defect and determine whether KB2115 acts locally or systemically to induce this effect; better knowledge of these influences would allow us to understand which TR isoforms and TR target genes are important and, possibly, to design ligands that would lack this harmful effect.
A future for thyromimetics?
While it will be difficult and possibly ill-advised to bring STRMs to the market as a long-term therapeutic for dyslipidemias or other aspects of metabolic disease, the fact remains that this class of ligands displays expected beneficial effects with greatly reduced harmful side effects (2). Thus, there could be alternate uses for this ligand class in cases in which it is important to modulate TH signaling pathways.
STRMs may be useful for several orphan indications. GC-1 and KB2115 reduce LDL cholesterol in mice devoid of LDL-R (82). It will be important to determine whether they act similarly in human homozygous familial hypercholesterolemia (106), which is caused by homozygous LDL-R mutations and leads to severe atherosclerosis for which therapies are limited. GC-1 has been designated as an orphan drug for adrenoleukodystrophy (Endochem Inc., California), caused by defects in an X-linked gene encoding a membrane transporter (ABCD1) leading to harmful accumulation of very long chain fatty acids. Since a related gene (ABCD2) is induced by T3 in liver, it is thought that ABCD2 induction could compensate for ABCD1 defects (107).
Despite problems in DITPA clinical trials, there are suggestions about possible uses for this compound and some may be applicable to other STRMs. While studies of DITPA effects in animal postinfarction models have yielded contradictory results, DITPA reduces pathologic remodeling and improves cardiac output postmyocardial infarction and acute administration reduces infarct size and attenuates early inflammatory response (108). Thus, DITPA could improve outcomes after myocardial infraction. Others have suggested applications for the TRβ-selective ligands in combating phenotypes of resistance to thyroid hormone syndrome (109). Finally, DITPA does not depend upon MCT8 for transport into neurons and it is proposed that DITPA or similar thyromimetics could affect partial rescue of neural phenotypes associated with inherited genetic defects in this gene (110).
Our own opinion is that STRMs would be most useful for short-term treatments of morbidly obese patients. Based on preclinical animal studies, we predict that GC-1 extrahepatic actions should induce fat burning in humans and that treatment durations required to obtain significant weight loss should be short enough to avoid harmful side effects, including those on cartilage, which only emerged after 1 year. We have been able to safely manage GC-1–dependent weight loss in rodents for periods of up to 60 days and have used GC-1 to effectively normalize body weight in several different obese mouse models, including C57BL/6 mice with diet-induced obesity and ob/ob mice fed a high-fat diet. In the latter case, we observed up to 20% reductions in total body fat over 3–4 weeks relative to a 5% fat gain for untreated control mice in the same period. When clinical guidelines recommend that patients in need of organ transplants are not eligible for surgery because of weight limits, we believe that similar degrees of managed fat loss could literally be lifesaving. It would obviously be crucial to perform such interventions in conditions in which patients were closely monitored for excessive increases in body temperature and other harmful effects. Based on these considerations, it is very important to explore possibilities for STRM-dependent weight loss in primate models or humans.
Footnotes
Acknowledgments
We would like to acknowledge Aijun Zhang and Carly Filgueira (Houston Methodist) for help with the figures.
Disclosure Statement
No competing financial interests exist.