
Abstract
Background:
Pituitary and thyroid autoimmunity can be triggered by pregnancy. We report the first association of combined growth hormone (GH) and prolactin secretion deficiency due to autoimmune damage to GH- and prolactin-secreting cells in a patient with postdelivery lactation failure, presenting subsequently with primary autoimmune hypothyroidism.
Patient Findings:
A 34-year-old woman presented with lactation failure following the delivery of her first child. She had a family history of hypothyroidism without a history of pituitary dysfunction. Physical examination did not show any abnormal findings. Laboratory investigations showed normal gonadotropin levels after the restoration of normal menstrual cycles following pregnancy, normal basal and stimulated cortisol levels, but an impaired GH response to insulin-induced hypoglycemia, and low basal prolactin and insulin-like growth factor-1 concentrations. Thyroid function was normal when initially investigated three months after delivery, but five months later, marked primary hypothyroidism (thyrotropin levels >100 mIU/L) occurred. Immunological investigation revealed the presence of antipituitary antibodies, identified by double immunofluorescence and targeting GH- and prolactin-secreting cells. Antithyroid antibodies, in the normal range three months postpartum, became significantly elevated when the hypothyroidism appeared. Autoimmune hypophysitis is responsible for selective or multiple pituitary-hormone deficiencies, sometimes involving thyrotropin secretion and causing secondary hypothyroidism, but usually associated with hyperprolactinemia. To our knowledge, this is the first observation of autoimmune hypopituitarism involving deficient growth hormone and prolactin secretion in a patient with lactation failure after delivery, subsequently followed by severe primary autoimmune hypothyroidism, thus falling into an unusual constellation of autoimmune polyendocrine syndrome type 3.
Conclusions:
Considering the well-known relationship between pregnancy and autoimmunity, an early postdelivery immunological and functional investigation in women presenting with disorders of lactation may be useful to detect potential pituitary and thyroid dysfunction even at a subclinical stage.
Introduction
Here, we describe a 34-year-old woman who presented with lactation failure after delivery, growth hormone (GH) and prolactin deficiencies, and subsequently severe primary autoimmune hypothyroidism. Immunological studies using four-layer double immunofluorescence indicated the presence of APA targeting GH- and prolactin-secreting cells, and elevated levels of antithyroglobulin and antiperoxidase antibodies at the subsequent appearance of thyroid dysfunction. To our knowledge, this is the first observation of autoimmune hypopituitarism involving GH and prolactin secretion in a patient with lactation failure after delivery, subsequently followed by severe primary autoimmune hypothyroidism, thus falling into an unusual constellation of autoimmune polyendocrine syndrome (APS) type 3.
Patient
A 34-year-old woman presented with failure of lactation following delivery of her first child. During the last trimester of pregnancy, she suffered from headaches, nausea, and vomiting. Her pregnancy progressed very well, and delivery occurred without complications. She had a previous history of polycystic ovary syndrome with endometriosis but without problems conceiving. The clinical history did not suggest pituitary dysfunction, although there was a family history of hypothyroidism.
Physical examination showed no apparent abnormal findings: she had normal visual fields and normal optic fundi, normal breasts, absence of galactorrhoea, and a normal body mass index (BMI; 20.3 kg/m2).
Laboratory investigations showed low basal levels of serum prolactin (<30 mIU/L, normal 40–530), and a low serum insulin-like growth factor-1 (IGF-1 73 μg/L, normal for age 109–424). Her serum cortisol level was normal (571 nmol/L, normal 200–700), and a fluid deprivation test did not show any impairment of her vasopressin synthesis and secretion. During insulin-induced hypoglycemia (glucose nadir 0.8 mmol/L), GH levels failed to increase normally (initial peak of 9.6 μg/L at the beginning but with a fall to 4.4 μg/L corresponding to the glucose nadir), while serum cortisol levels increased normally (1152 nmol/L, normal >550). Gonadotrophin concentrations were normal after the restoration of normal menstrual cycles three months after delivery, suggesting a normal function of her hypothalamic–pituitary–gonadal axis. Her thyroid function was normal when initially investigated three months after delivery, but five months later, she developed marked primary hypothyroidism with a low serum free T4 <5.0 pmol/L (normal 10–24) and significantly raised thyrotropin levels (>100 mIU/L, normal 0.4–4.0). Thyroid ultrasound findings were suggestive of thyroiditis. Antithyroid antibodies, which were in the normal range at the first evaluation, showed a significant increase (antiperoxidase 349 IU/mL, antithyroglobulin 86 IU/mL; normal <60 for both antibodies).
MRI of the hypothalamus–pituitary region performed at presentation and six months later was normal on both occasions. APA were detected by simple indirect immunofluorescence method on cryostat sections of young baboon pituitary glands supplied by Halifax spa (Polverara, Pordenone, Italy) as previously described (8,9). In particular, fluorescein isothiocyanate (FITC)-conjugated goat anti-human Ig was used to detect the presence of APA. Taking into account the sensitivity and specificity (low) of the immunofluorescence method, we usually use not only a cut-off of 1:8 to consider the sera processed by this method as positive, but also a particular immunofluorescence pattern to improve the specificity and the sensitivity of the method, namely an immunostaining involving some but not all the pituitary cells (9). APA were present in our patient at a high titer (1:128). Subsequently, the same serum was retested by four-layer double immunofluorescence to characterize the pituitary cells targeted by APA. In particular, after the first immunostaining step, using sera of a positive patient and FITC-conjugated goat anti-human Ig, on the same pituitary cryostat section, a second immunostaining step using rabbit antisera to corticotropin (ACTH), GH, thyrotropin (TSH), prolactin, luteinizing hormone (LH), and follicle-stimulating hormone (FSH) (Histo-line Srl, Milano, Italy), separately, followed by rhodamine anti-rabbit Ig, was performed. The different color of anti-Ig conjugate against the human serum and against the animal serum, respectively green (FITC) and red (rhodamine), allowed the direct assessment of whether the patient's serum and the animal's serum stained the same or different pituitary cells (10).
The use of a four-layer immunofluorescence method in the second sandwich rabbit serum using the abovementioned antisera demonstrated that APA were directed against prolactin- and GH-secreting cells, but not against TSH-, ACTH-, LH-, and FSH-secreting cells (Fig. 1).
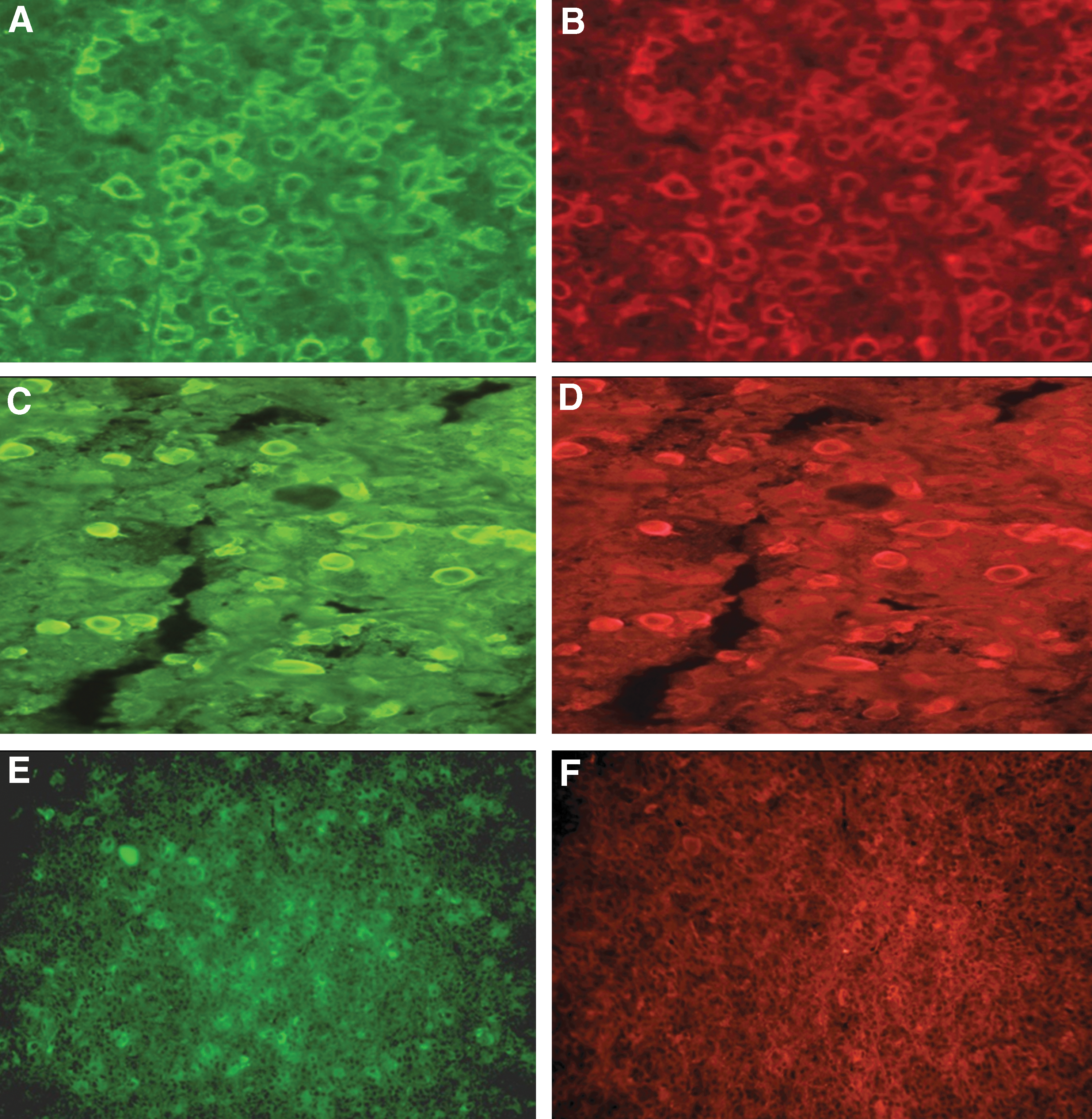
Characterization of antipituitary antibodies by double immunofluorescence in the patient serum: the correspondence between the cells colored in green in the first step
Discussion
LYH is an autoimmune disease of the pituitary gland, which can present with varying degrees of pituitary hormonal impairment and/or with symptoms related to pituitary enlargement. Organ-specific antibodies are good markers of many autoimmune endocrine diseases. However, the role of APA in LYH is still debated because of various methodological difficulties and uncertainties in the clinical interpretation. In fact, conflicting results in detecting APA using different methods (immunofluorescence, immunoblotting, and radioligand) led to uncertainties about the clinical relevance of these antibodies, particularly because the true pituitary antigens reacting with these antibodies are still unknown (5,11 –14). A recent revaluation of APA by an immunofluorescence method in a large population of patients with APS allowed the detection of patients with pituitary impairment in those who were APA positive at high titers, and showed a particular immunofluorescence pattern with an immunostaining involving some but not all pituitary cells (9), suggesting that the occurrence of LYH in patients with other autoimmune diseases and in patients with apparently idiopathic hypopituitarism is more frequent than so far considered (14). In particular, in patients with idiopathic GH deficiency, in those with idiopathic hypogonadotropic hypogonadism, and in those with ACTH deficiency, a four-layer immunofluorescence method showed that APA immunostained selectively GH-, FSH/LH-, or ACTH-secreting cells respectively, suggesting that these cells were the main target of these antibodies (5 –8). Following this, it was suggested that APA, when present at high titers and with a particular immunostaining pattern, may be a good diagnostic and predictive marker of autoimmune pituitary impairment even without a definite pathogenetic role in LYH.
With respect to the relationship between autoimmune diseases and pregnancy, it is well known that some autoimmune diseases, such as autoimmune thyroiditis, may improve during (15), while others, such as autoimmune myocarditis (16) and systemic lupus erythematosus (17), may worsen in pregnancy. LYH shows a close temporal relationship with pregnancy. In particular, LYH may be present in young pregnant women in the second or third rather than in the first trimester. Moreover, there is a frequent spontaneous remission of hypopituitarism and pituitary swelling with disappearance of APA after pregnancy, although in some cases a recurrence of LYH during long-term follow-up after delivery has been described (18). In particular, the search for APA in patients with Sheehan's syndrome (SS) many years after the onset of SS was performed to verify whether an autoimmune pituitary process could contribute to late hypopituitarism in this condition. Interestingly, antibodies to the pituitary gland were found in such patients, suggesting that the occurrence of an autoimmune process involving the pituitary gland may contribute to late pituitary dysfunction in these patients (19).
Currently, the causes of the particular relationship between LYH and pregnancy are still debated. However, it is well known that there is hypertrophy and hyperplasia of pituitary cells during pregnancy, and that hyperestrogenism increases pituitary blood flow. In this situation, it is possible that pituitary cryptic antigens, becoming much more accessible to the immune system, may trigger an autoimmune pituitary process. Another possible cause of the close association between LYH and pregnancy could be the presence of high prolactin levels in this condition. Some studies have shown an immunostimulatory role for prolactin; in particular, high prolactin levels could have a role in perpetuating the immune process in LYH (20,21). In fact, hyperprolactinemia is a common finding in patients with LYH. A multifactorial etiology related to the diffuse inflammatory process in the pituitary has been proposed for the hyperprolactinemia, namely the loss of the inhibitory effect of dopamine and an alteration of dopamine receptors, lactotrope hyperplasia, or escape of prolactin into the circulation secondary to massive cellular destruction (3,21). Only isolated cases of women with LYH have been described presenting with hypoprolactinemia and failure to lactate in the postpartum period (22 –24).
Our patient, after delivery, developed GH and prolactin deficiencies with lactation failure, in the presence of a normal thyroid function and no evidence of thyroid autoimmunity. Severe hypothyroidism appeared several months later, but it was not secondary to impaired TSH secretion (secondary hypothyroidism) due to combined pituitary dysfunction, as described in some APA positive patients with autoimmune hypopituitarism (25). In fact, the double immunofluorescence excluded that APA were directed also against thyrotrophs (Fig. 1). Instead, our patient showed severe primary hypothyroidism with appearance of elevated levels of antithyroglobulin and antiperoxidase antibodies, and sonographic findings of autoimmune thyroiditis. This association is certainly related to an autoimmune pathology, in which the pregnancy played a prevalent triggering role. To our knowledge, this is the first observation of GH and prolactin deficiency with presence of APA targeting GH- and prolactin-secreting cells in a patient with lactation failure after delivery. The association between primary hypothyroidism with antithyroid antibodies and pituitary autoimmune dysfunction with the presence of APA configuring the occurrence of autoimmune adeno-hypophysitis, indicates that this patient can be categorized as having APS type 3 (26 –28). Considering the well-known relationship between pregnancy and autoimmunity, an early postdelivery immunological and functional investigation in women presenting with disorders of lactation may be useful to detect potential pituitary and thyroid dysfunction even in a subclinical stage.
Footnotes
Author Disclosure Statement
No competing financial interests exist.