
Abstract
Background:
Ectopic thyrotropin (TSH)-secreting tumors are extremely rare. To our knowledge, only three cases have previously been reported so far, but the tumors were not studied ultrastructurally and in vitro. We present a case that was extensively examined to gain deeper insights in terms of the histopathological features and hormonal secretion profile of the tumor.
Patient Findings:
A 49-year-old female complained of nasal obstruction for 15 years and thyrotoxicosis for one and a half years. Except for a high basal TSH with concomitantly elevated free tri-iodothyronine (FT3) and free thyroxine (FT4) levels, her pituitary hormone profile yielded normal results. Magnetic resonance imaging revealed a 2 cm×2 cm mass in the nasopharynx, which showed an increased tracer uptake on octreotide scintigraphy. Preoperative treatment with octreotide effectively reduced serum TSH, FT3, and FT4 to normal levels. The mass was endoscopically removed via an endonasal approach. Immunophenotyping and hormone determination of cultured cells confirmed that the mass was a plurihormonal TSH-/growth hormone (GH)–/prolactin (PRL)-producing adenoma. Co-expression of TSH and GH was found in most cells. Electron microscopy showed that the adenoma was formed by a single cell type, with secretory granules of small size. In vitro studies demonstrated that octreotide reduced both TSH and GH secretion.
Summary:
We report an ectopic TSH-secreting tumor, which had plurihormonal secretion in vitro, including TSH, GH, and PRL. Histologically, it mimicked a TSH-secreting pituitary adenoma. Octreotide was useful in the diagnosis and treatment of this ectopic TSH-secreting tumor.
Conclusions:
Ectopic TSH-secreting tumors are extremely rare. In terms of hormone secretion profile, histological characteristics, and response to octreotide, they are similar to pituitary TSH-secreting adenomas, suggesting that they are of identical cell origin.
Introduction
Herein, we reported a fourth patient presenting with an ectopic TSH-secreting tumor—a woman with hyperthyroidism caused by a nasopharyngeal tumor. Furthermore, we intensively examined the tumor, both in vivo and in vitro, to gain more insights in terms of the histopathological features and hormonal secretion pattern of the tumor.
Patient
A 49-year-old woman was referred to the PUMC hospital in July 2012 because of nasal obstruction for 15 years and thyrotoxicosis for one and a half years. In 1998, at the age of 34, the patient began to suffer from progressively exacerbating nasal obstruction. She was suspected of having sinusitis in a local hospital, and no other examination was conducted at that time. One and a half years ago, the patient began to have palpitations, sweating, nervousness, and weight loss. At that time, thyroid function tests showed a high FT4 (42.1 pmol/L; normal 7.9–14.4), FT3 (14.7 pmol/L; normal 3.8–6), and serum TSH (5.9 mIU/L; normal 0.34–5.6). An electrocardiogram showed atrial fibrillation and a thyroid ultrasound revealed a diffuse enlargement of the thyroid gland. She was diagnosed with Graves' disease and was given methimazole (30 mg daily) and betaloc. The patient took methimazole irregularly, and her symptoms showed no obvious improvement. Further inquiry revealed no family history of thyroid dysfunction.
On admission, physical examination showed a diffuse enlargement of the thyroid gland and an irregular heart rate of 90 beats per minute. No ophthalmopathy, pretibial myxedema, or signs of acromegaly were found. Thyroid function test showed a high basal TSH level (8.3 mU/L; normal 0.38–4.34) together with elevated FT3 (18.6 pmol/L; normal 2.7–6.3) and FT4 (54.6 pmol/L; normal 10.4–24.3). The glycoprotein hormone α-subunit was not measured. Antithyroperoxidase, antithyroglobulin and anti-TSH receptor autoantibodies were negative. Random serum growth hormone (GH, 1.3 ng/mL; normal <2.0), serum insulin-like growth factor 1 (IGF1, 109 ng/mL; normal 94–252) and prolactin (PRL, 8.58 ng/mL; normal 3.5–10.5) levels were normal. No abnormality of the gonadal axis or the adrenal axis was detected. Nasal endoscopy exhibited a nasopharyngeal mass on the posterior portion of the midline septum. Magnetic resonance imaging (MRI) showed a Rathke cleft cyst in the pituitary gland and a 2 cm×2 cm mass in the nasopharynx. A computed tomography (CT) scan confirmed that the lesion did not involve the local bones. Scanning with 99mTc-labeled octreotide showed an increased tracer uptake of the mass, suggesting a tumor of neuroendocrine nature (Fig. 1A–C).
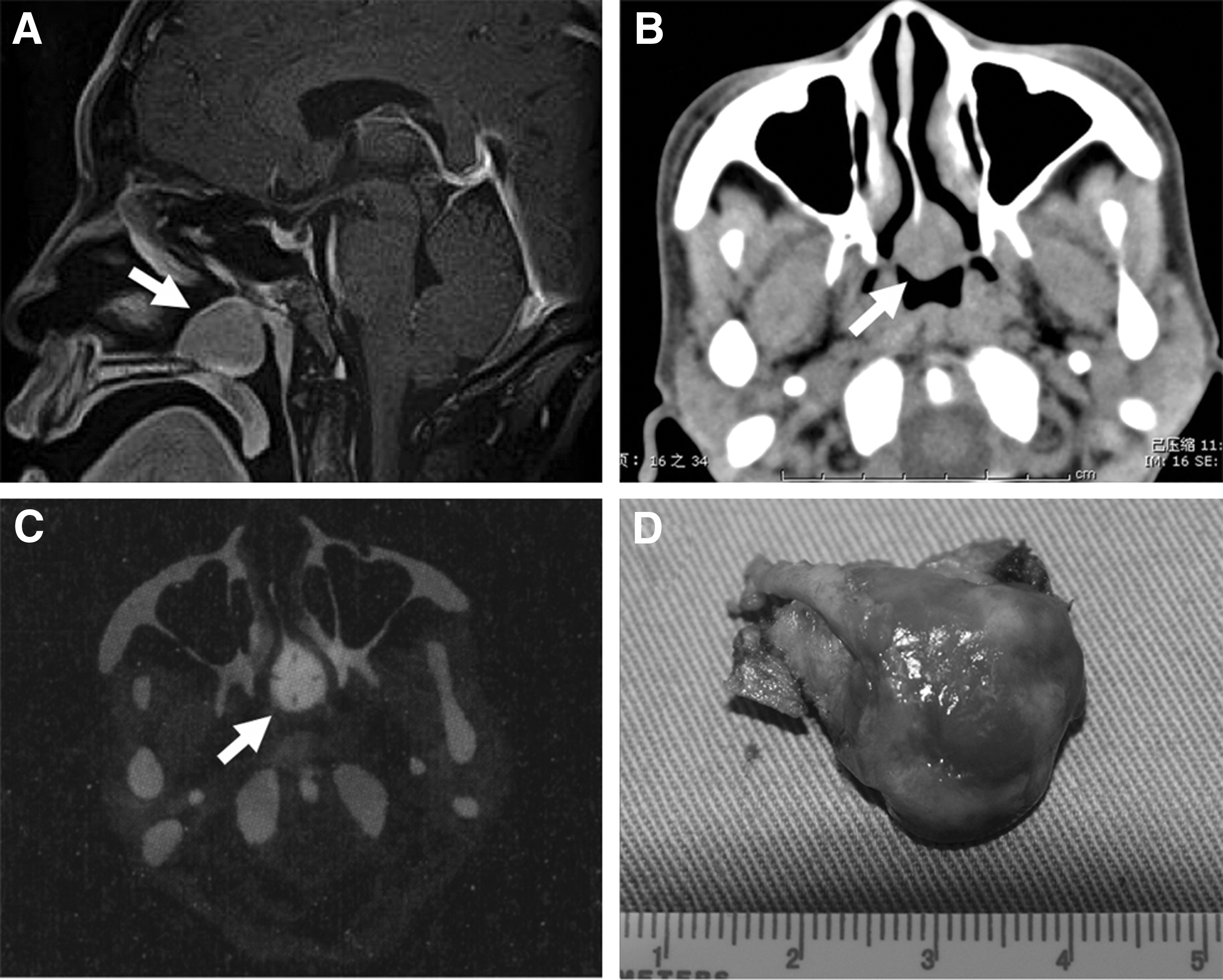
Magnetic resonance imaging (MRI)
On the basis of the aforementioned findings, the diagnosis of an ectopic TSH-secreting tumor in the nasopharynx was tentatively established, thereby explaining the nasal obstruction and the thyrotoxicosis of the patient. An octreotide suppression test (0.1 mg, subcutaneously, every 8h) was performed. After treatment with octreotide for one day, TSH decreased into the normal range, and FT3 and FT4 levels declined to the reference range at day 7 after octreotide administration (Fig. 2). Then, the mass was endoscopically removed via an endonasal approach. The gross pathological examination demonstrated a well-encapsulated red solitary lesion measuring 2 cm in diameter (Fig. 1D). The patient discontinued the octreotide treatment postoperatively, and had a normal thyroid function on follow-up examination. She gave her informed consent for the use of the tumor material for research purposes. The investigation was approved by the local ethical committee.
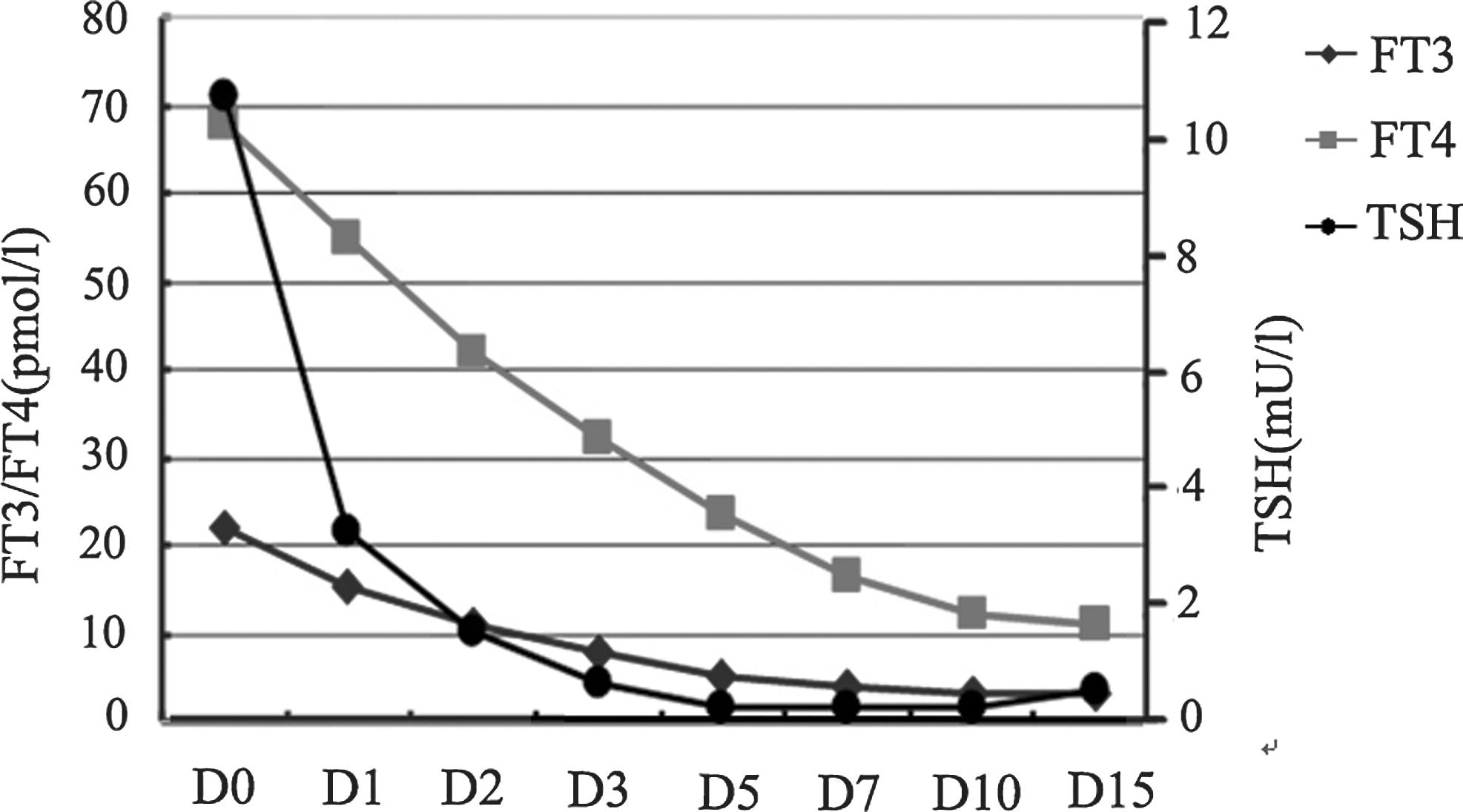
Serum FT3, FT4, and thyrotropin (TSH) levels in the octreotide suppression test. Reference ranges: TSH 0.38–4.34 mU/L; FT3 2.7–6.3 pmol/L; FT4 10.4–24.3 pmol/L.
Histologically, the tumor was composed of monomorphic cells with a distinct cell border, abundant granulated cytoplasm, and round or oval nuclei. Relatively large amounts of blood sinuses existed in the tumor (Fig. 3C). Immunohistochemistry showed strong immunoreactivity for chromogranin, synaptophysin, TSH, GH, and vascular endothelial growth factor; sporadic immunoreactivity for PRL; and no evidence for the expression of follicle-stimulating hormone (FSH), luteinizing hormone (LH), adrenocorticotropin (ACTH), β-human chorionic gonadotropin (β-HCG), P53, erb-B2, and epidermal growth factor receptor. The Ki-67 index was <2% (all the above primary antibodies were from Dako). Consecutive 3 μm sections revealed that most tumor cells were positive for both TSH and GH (Fig. 3A, B). Electron microscopy showed that the adenoma consisted of monomorphous cells (Fig. 3D). Unlike typical thyrotrophs, which are predominately elongated or polyhedral with long cytoplasmic processes, the cells of this tumor were spherical or oval. The nuclei were of spherical, oval, or irregular shape and predominantly contained nucleoli. Some cells had two nuclei. The cytoplasm contained relatively large amount of mitochondria and lysosomes. Numerous secretory granules were scattered across the cytoplasm or lining the cell membrane. They were similar in size, shape, or electron density with a diameter of 60–120 nm. The cells contained no GH-type large secretory granules or fibrous bodies.
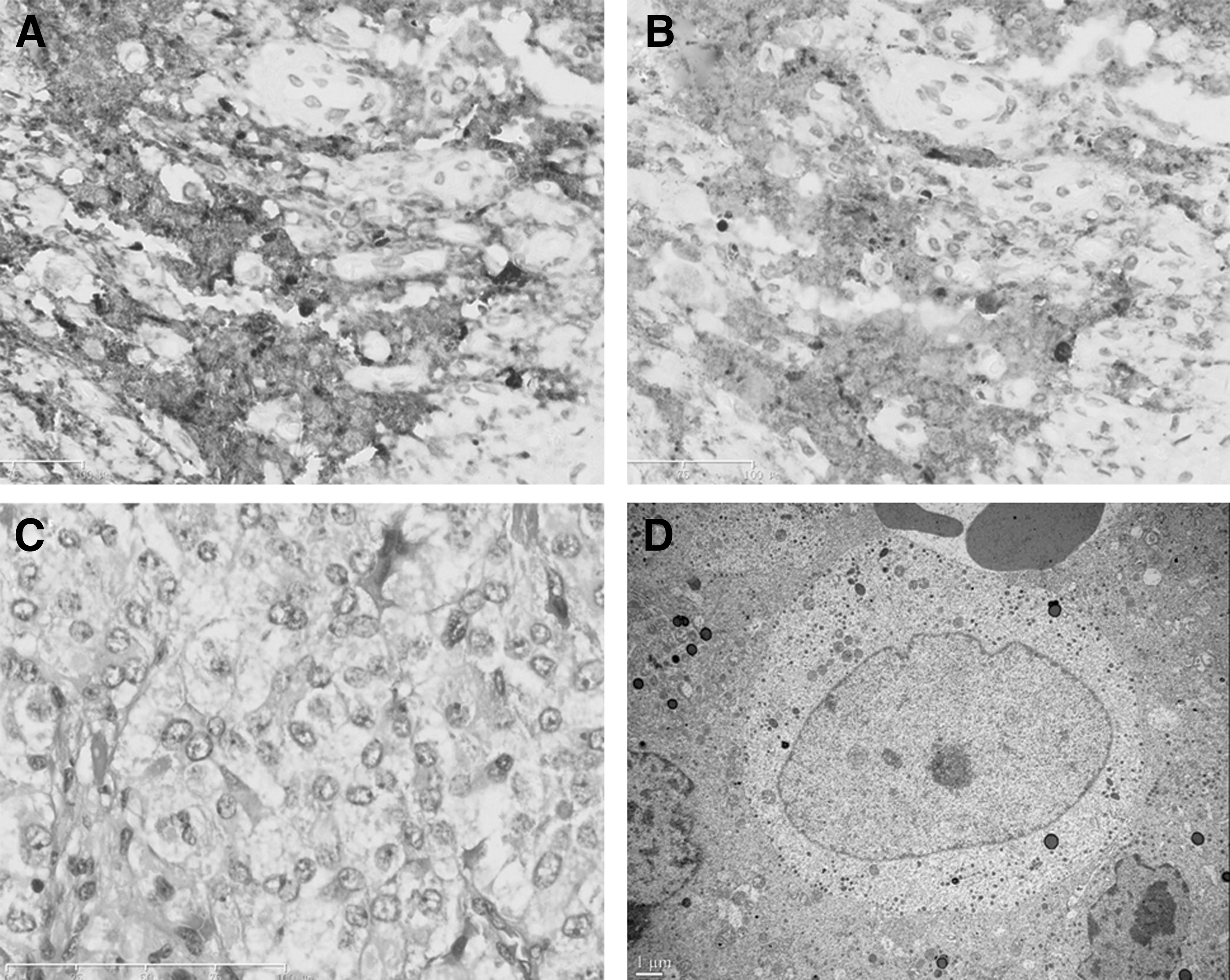
Immunostaining for TSH
Surgically removed tumor tissues were put into Dulbecco's modified Eagle's medium (DMEM) for secretion studies, as previously described (6). Briefly, the tissue sample was cut into 1 mm3 fragments. Six pieces were incubated in 1 mL DMEM containing HEPES at 37°C in a shaking bath. After 90 min, the medium was collected and the fragments were incubated with fresh medium for 30 min. Media were collected and centrifuged for measurement of pituitary hormones. TSH (2,150 mU/L), GH (2,330 ng/mL), and PRL (16.2 ng/mL) were detected in the medium of the first 90 min incubation, and only TSH (125 mU/L) and GH (389 ng/mL) could be observed in the medium of 30 min incubation. No other pituitary hormones, such as ACTH, FSH, and LH, were detected.
Primary culture of the tumor cell was conducted. Tumor tissue (800 mg wet weight) was washed three times with phosphate-buffered saline (PBS), cut into small pieces, and dispersed by incubation in PBS containing 0.35% collagenase I for 20 minutes at 37°C. Dispersed cells were harvested by centrifugation, washed once, and subsequently resuspended in culture medium (DMEM containing 10% fetal calf serum, 25 mM HEPES, 100 μg/mL penicillin, and 100 μg/mL streptomycin). The cells were plated into 24-well plates coated with poly-

In vitro studies. Phase-contrast microscopy
Octreotide suppression test was conducted at day 5. The cells were incubated in fresh culture medium alone (controls) or in culture medium containing 1 μM octreotide (Novartis) for 4 hours. For each group, four different culture wells were tested. At the end of the experiment, the media were collected, centrifuged, and stored at −20°C until measurement of TSH and GH. The TSH and GH levels in the octreotide-treated group were 12.6±1.5 mU/L and 6.0±1.6 ng/mL respectively, compared to 22.2±2.5 mU/L and 11.2±1.6 ng/mL in the control group. The difference between the two groups (t test by employing SPSS V13.0) was statistically significant (p<0.05 for both).
Discussion
Only three cases of ectopic TSH-secreting tumor have been reported before, indicating that this is an extremely rare entity (Table 1) (3 –5). The previously reported patients and the patient presented here had an ectopic tumor in the nasopharyngeal cavity. Except for the distinct finding of a stuffy nose, ectopic TSH-secreting tumors share the same symptoms and biochemical alterations as pituitary TSH-omas. In terms of elevated circulating thyroid hormones with inappropriately normal or elevated TSH concentrations, the biochemical findings associated with central hyperthyroidism, TSH-omas are not readily distinguishable from resistance to thyroid hormones (RTH). Several diagnostic steps have to be taken to differentiate a TSH-oma from RTH. Apart from imaging examinations such as MRI and CT, which can show pituitary or ectopic masses, an unbalanced hypersecretion of the pituitary glycoprotein hormone alpha-subunit (α-GSU) and an elevated α-GSU/TSH molar ratio are suggestive for the presence of a TSH-oma (1). However, in our patient α-GSU was not measured. Since most pituitary TSH-oma cells have somatostatin receptors and remain sensitive to somatostatin and its analogues, radionuclide-labeled octreotide scintigraphy and an octreotide suppression test may be of great help for establishing the diagnosis. In the present case, in vitro studies on primary tumor cell cultures showed that octreotide could inhibit TSH secretion, and that the inhibitory effect was positively correlated with the response observed in vivo. This evidence, together with the in vivo receptor imaging study, suggest that this ectopic TSH-secreting tumor had somatostatin receptors.
ACTH, adrenocorticotropin; F, female; FSH, follicle-stimulating hormone; GH, growth hormone; LH, luteinizing hormone; M, male; ND, not described; PRL, prolactin; TSH, thyrotropin; +, positive; −, negative.
Immunocytochemical staining and in vitro hormone secretion studies showed that this ectopic TSH-secreting tumor is of plurihormonal nature and secretes TSH, GH and PRL. Two previously reported ectopic TSH-secreting tumors were also immunologically considered to be plurihormonal adenomas (3,5). However, in these reports, no in vitro studies had been conducted to confirm the secretion of various hormones. With pituitary TSH-omas, co-secretion of GH and PRL is found in about 30% of the cases. Up to 16% of the patients co-secrete GH and 11% PRL, while co-secretion of other pituitary hormones is rare. An even higher incidence of expression of GH and PRL in TSH-omas has been detected by immunohistochemical studies (7 –9). In our case, immunocytochemistry revealed colocalization of TSH and GH in most tumor cells, indicating these two hormones were expressed mainly by the same cells. In vivo, however, only TSH was elevated whereas GH, IGF1, and PRL were normal. Ultrastructurally, the monomorphic cellular appearance and the single type of secretory granulations, similar to the one found in normal thyrotroph cells, suggested that TSH and GH may came from the same secretory granules, as reported in several cases of pituitary adenomas (10,11). Malarkey et al. reported a case of a pituitary adenoma co-secreting TSH and GH, which presented with acromegaly and hyperthyroidism (11). Immunoelectron microscopy localized the two biochemically distinct peptides in the same cell type, often in the same secretory granules. Moreover, other morphologic features of TSH- and GH-secreting adenomas have been described. Ozawa et al. reported a TSH- and GH-secreting pituitary adenoma that was ultrastructurally monomorphous, but immunohistochemically polymorphous (12). By electron microscopy, the adenoma was found to be composed of a single cell type resembling thyrotrophs, and did not have any characteristics of somatotrophs. But immunohistochemical studies showed positive staining for TSH and GH in different cells, not in the same cells. In addition, several papers described adenomas consisting of two distinct cell types, thyrotroph-like cells containing TSH and somatotroph-like cells containing GH (13,14). The characteristic concomitant hypersecretion of TSH and other pituitary hormones in this ectopic tumor has been reported in many pituitary adenomas, suggesting all these tumors derived from an identical cell type, which supports the assumption that ectopic tumors arise along the migration path of the Rathke's pouch or from aberrant anterior pituitary cells within the pituitary stalk or pars tuberalis (4).
Detection of TSH and GH in culture media proved that these hormones were secreted, but the patient only exhibited increased serum TSH level and her circulating GH and IGF1 were in the normal range. Moreover, tissue incubation revealed a relatively high GH release, suggesting that the tumor had a strong GH secretion. With most plurihormonal pituitary adenomas, hormonal secretions in vitro are closely associated with biochemical changes and the clinical presentation of the patients (15,16). Inconsistent findings between the pituitary hormone secretion in vivo and in vitro have been previously reported (10,17). Bertholon-Gregoire et al. reported an elevated GH release rate (100 ng/day) in the culture medium of a pituitary TSH-oma, but the circulating GH level of the patient was found to be normal (10). Aylwin et al. also reported three GH-secreting adenomas that released high levels TSH in vitro (17). However, clinically the patients presented with acromegaly alone, but no signs of hyperthyroidism. The reasons for this inconsistency remain unclear. In the current study, tissue incubation soon after surgery showed abundant GH release into the medium, suggesting that GH was synthesized and may perhaps have been released in vivo. Whether the GH release was just limited to the tumor and did not enter the circulation, or whether the GH secretion was too low to yield appreciable biological changes or clinical signs warrants further investigation.
In summary, we describe a case of an ectopic TSH-secreting tumor in the nasopharyngeal cavity presenting with nasal obstruction and thyrotoxicosis, which in vitro was proved to be a plurihormonal adenoma. Its histology and ultrastructure were similar to those of pituitary adenomas suggesting it had an identical cellular origin. Octreotide was useful in the diagnosis and treatment of this ectopic TSH-secreting tumor.
Footnotes
Acknowledgments
We are indebted to Dr. Ping Li and Dr. Xiaohu Shi for their help in the treatment of the patient, and to Dr. Dachun Zhao, Mrs. Shuangyu Lu, Mrs. Yingying Hu, Mrs. Peng Yang, and Mr. Wei Dai for their technical assistance.
Author Disclosure Statement
The authors declare that no competing financial interests exist.