
Abstract
Background:
Medullary thyroid cancer, although most commonly sporadic, may be part of the multiple endocrine neoplasia type 2 (MEN2) syndromes, generally due to mutations in the RET proto-oncogene. The majority of these mutations are located in exons 10, 11, and 13–16. More rarely, mutations in other exons have been described. We report for the first time a family from the United States with a rare mutation involving exon 8 of the RET proto-oncogene, corresponding to a p.Gly533Cys substitution (G533C) leading to the development of MEN2A syndrome in several affected family members. This mutation had only been previously described in a large family in Brazil and in 7.75% of patients with apparently sporadic medullary thyroid cancer (MTC) in Greece.
Methods:
Given a strong index of suspicion, a genetic analysis to evaluate for uncommon mutations in the RET proto-oncogene identified the presence of the G533C missense mutation, despite initial negative screening for common mutations. We describe a family with a total of 47 individuals from five generations with multiple members affected with this mutation.
Results:
Our data suggest that in patients with this mutation, pheochromocytoma is more common than previously reported, and that in some cases this mutation may be associated with a more aggressive phenotype than initially described.
Conclusions:
MEN2A due to the G533C mutation in exon 8 may be more common and more aggressive than previously recognized. In patients with medullary thyroid cancer with negative screening for common mutations in the RET oncogene but a strong index of suspicion, DNA sequence analysis of less commonly involved exons should be considered.
Introduction
M
Activating germline mutations of the RET proto-oncogene, located on chromosome 10q11.2, are associated with the described FMTC, MEN2A, and MEN2B hereditary MTC syndromes, with the majority of these mutations located in exons 10, 11, and 13–16. In more than 90% of cases of MEN2A, RET mutations are present in exons 10 and 11. Rarely, other exons may be involved, and mutations have been reported in isolated families. However, a few families have been reported without demonstrable mutations (3).
In this manuscript, we describe, for the first time, a family from the United States with a rare mutation in exon 8 of the RET proto-oncogene, c.1597G>T, resulting in a p.Gly533Cys substitution (G533C). This mutation had only been previously described in a large family in Brazil and two unrelated families from Greece (4,5).
Subjects and Methods
The pedigree of this family is reported in Figure 1. A total of 47 individuals from five generations were reported by the proband and subsequent family members seen at the Mayo Clinic. The index case (III-2) is a 51-year-old Caucasian woman from the United States, diagnosed with a thyroid nodule during routine physical examination. Ultrasound examination demonstrated multiple bilateral thyroid nodules, some of which were calcified, with the largest one in the right lobe measuring 1.5 cm in greatest dimension and the largest one in the left lobe measuring 2 cm in largest dimension. Fine-needle aspiration biopsy of the dominant nodules in both lobes was performed with cytology consistent with MTC. Basal serum calcitonin (CT) was elevated at 707 pg/mL (normal <8 pg/mL). Serum calcium and PTH were normal, and screening for pheochromocytoma with 24-hour urine for catecholamines and metanephrines was negative.
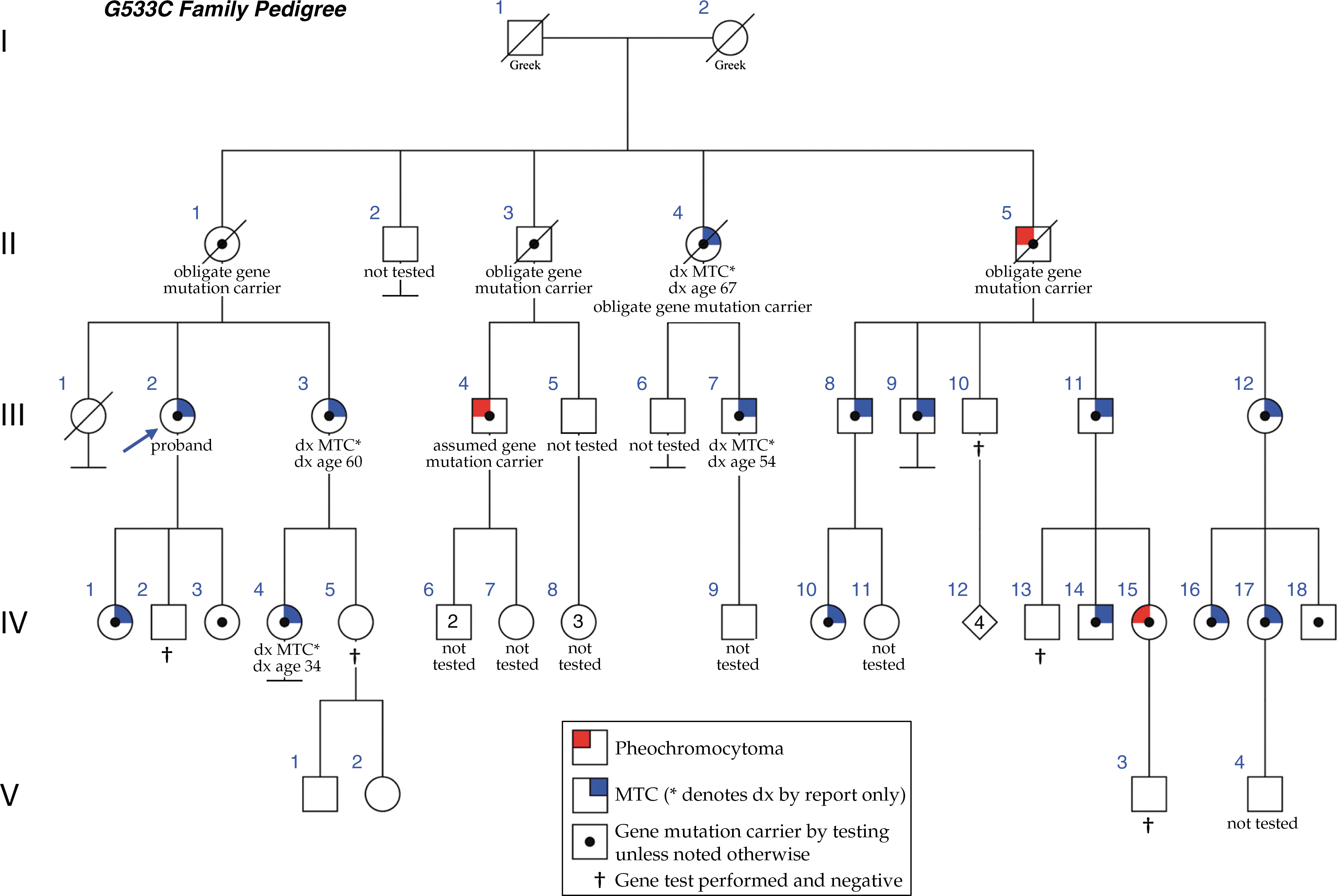
Pedigree of family with G533C mutation. Affected individuals are designated by the symbols shown in the key at the bottom of the figure. The proband is identified with an arrow.
Our patient underwent thyroidectomy and central compartment (level VI) lymph-node dissection. Final histopathology demonstrated multifocal (MF) bilateral (BL) MTC with the largest tumors in the right and left lobe each measuring 1.6 cm in greatest dimension (Fig. 3). One out of seven resected lymph nodes was positive for metastatic MTC. Basal serum CT measured two months after surgery was undetectable (<5 pg/mL). Within one month of her surgery, one of her sisters was also diagnosed with, and underwent surgery for, MTC.
A comprehensive family history was obtained from the proband at the time of genetic counseling. A total of 16 members of this family have tested positive for the G533C mutation. An additional five individuals are presumed mutation carriers either because they have children who have tested positive, or because they had a MEN2-associated diagnosis (MTC or pheochromocytoma). Four of these patients have passed away (II-1, II-3, II-4, and II-5), and another one is still alive (III-4) and was diagnosed with a pheochromocytoma at the age of 29, but declined further evaluation for MTC or genetic testing. Thus, 21 mutation carriers have been identified to date. This includes three diagnoses of pheochromocytoma. The first diagnosis was reported in a maternal uncle now deceased (II-5), who had undergone a right adrenalectomy at the age of 65 years for a 10.5×9.5×7 cm pheochromocytoma. Review of his outside medical records confirmed this diagnosis. The second diagnosis occurred in a first cousin (III-4) who presented with severe hypertension at the age of 29 and underwent a resection of a 6×3 cm pheochromocytoma 10 years before at the Mayo Clinic. A third diagnosis of pheochromocytoma was made in a 29-year-old woman (IV-15) after she underwent genetic testing for the familial mutation. Even though she was asymptomatic and normotensive, screening with 24-hour urine catecholamines and metanephrines showed very high total metanephrine levels of 4406 μg/24 hours (normal 142–510 μg/24 hours). An abdominal computed tomography (CT) scan confirmed the presence of a 5 cm pheochromocytoma (Fig. 2). She underwent an uncomplicated laparoscopic adrenalectomy, and a 5.2×4.1×2.7 cm pheochromocytoma was resected. Thyroidectomy in this patient showed focal C-cell hyperplasia but no MTC (Fig. 3).
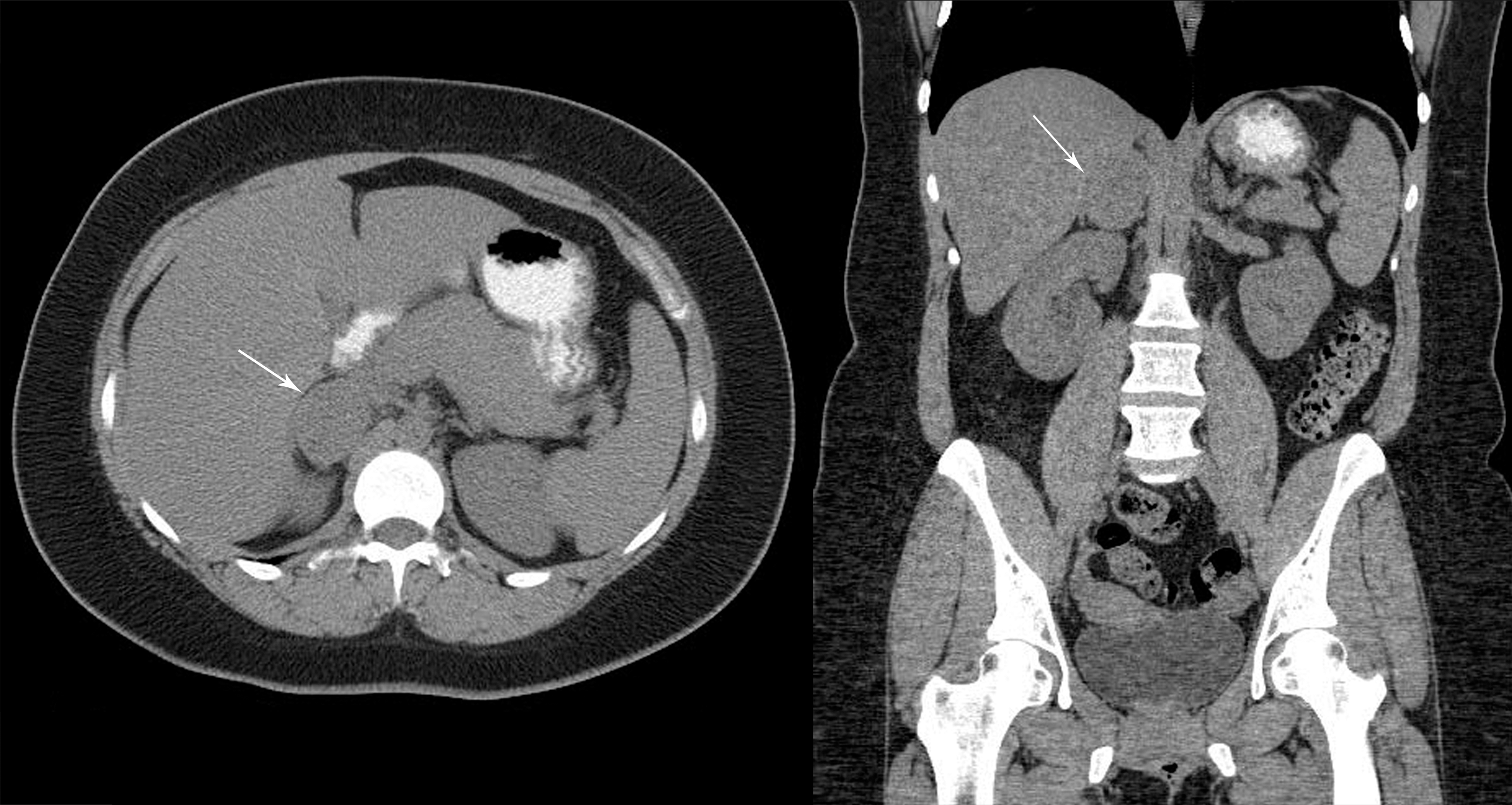
Computed tomography scab of the abdomen of asymptomatic mutation carrier (IV-15) with 5 cm right adrenal pheochromocytoma discovered during preoperative screening for thyroidectomy.

Histological sections from proband (III-2).
Twelve of the 16 known gene carriers were evaluated by the endocrinology department at the Mayo Clinic, and underwent laboratory testing to include serum basal CT measurement, testing for serum calcium, PTH, 24-hour urine collection for measurement of catecholamines and metanephrines, and thyroid ultrasound examination. Family members seen by the endocrinology department were referred to medical genetics for genetic counseling.
The study was approved by the Institutional Review Board of the Mayo Clinic. Table 1 summarizes our findings.
Numbers in parentheses in histopathology represent numbers of tumors found. aRefers to diagnosis of primary manifestation. MTC, medullary thyroid cancer; CT, calcitonin; HPT, hyperparathyroidism; BL, bilateral; MF, multifocal; TN, thyroid nodule(s); R, right; L, left; CCH, C-cell hyperplasia; N/A, not available.
Results
RET genetic analysis
Initial DNA sequence analysis in the index subject was performed at Mayo Medical Laboratories to test for the presence of a mutation in exons 10, 11, 13, 14, 15, and 16 of the RET proto-oncogene. A mutation was not detected. However, given a strong index of suspicion because of the reported family history, a DNA sample was submitted to an outside laboratory (GeneDx) for evaluation of less commonly reported mutations.
Bidirectional sequence analysis of exons 1–9, 12, and 17–20 of the RET gene and the flanking intron/exon boundaries revealed the presence of the G533C missense mutation, which has been previously reported in the literature (4,5). After this mutation was identified in the index patient, all other members of this family could be tested for this specific mutation, which was subsequently identified in 16 out of 21 relatives tested. Five individuals in this family have tested negative for the mutation, thus far.
With regard to other tests, all patients evaluated at the Mayo Clinic had serum calcium levels measured, and in all except one patient, these were normal. One patient had an elevated serum calcium level, but PTH measured in the same sample was appropriately suppressed. None of the patients in our cohort was found to have evidence of primary hyperparathyroidism.
Discussion
In 2003, Da Silva et al. reported for the first time a novel missense mutation in exon 8 in the cysteine-rich domain of the RET proto-oncogene, resulting in substitution of glycine 533 by cysteine (G533C) in a six-generation family from Brazil (4). This mutation is defined at the nucleotide level as c.1597G>T. Affected members of this six-generation family (76 out of 229 subjects) presented with isolated FMTC. Since then, this G533C mutation has been reported in a total of four additional unrelated families from Greece (5 –7). In two of these families, affected members were found to have isolated FMTC (6). However, in 2007, Bethanis et al. reported for the first time the case of a patient affected with this mutation presenting with bilateral pheochromocytomas as the initial manifestation that led to the diagnosis of MEN2A (7). Since then, two additional unrelated Greek patients were reported in whom pheochromocytoma was the initial presenting feature, indicating that these families had MEN2A rather than FMTC (5,7). Furthermore, eight years after the initial report by Da Silva et al. of this novel mutation, these authors reported the first case of pheochromocytoma in a mutation carrier in the Brazilian family initially described (8), and they demonstrated via in vitro studies the pathogenicity of this mutation, which seems to confer a malignant phenotype to PCCL3 cells (8). In 1999, Pigny et al. reported a family in which a novel nine-base pair insertion in exon 8 of the RET oncogene resulted in an additional cysteine residue; four affected members of this family had the FMTC phenotype (9).
More recently, Sarika et al. evaluated 129 unrelated Greek patients who had been diagnosed with “sporadic” MTC (all of them had negative family history, no other features of MEN2A or MEN2B, and tested negative for mutations in exons 10, 11, 13, 14, and 15). They found that 10 of these patients (7.75%) were positive for the G533C mutation (10), indicating that this mutation is more common than previously recognized in the Greek population, increasing the prevalence of the inherited form of MTC at their institution from 27.9% to 33.5%, one of the highest reported so far (10,11). The mean age of these patients at presentation was 51.8 years. Of these patients, three had locoregional metastatic disease. When 23 family members of affected patients were evaluated, 14 were also found to be carriers of this mutation (10). Because of the relatively high prevalence of this mutation in this Greek population, the authors are currently examining the possibility of a founder effect, as these apparently unrelated families come from two specific areas in rural Greece. Given these findings, the authors have recommended that evaluation for mutations in exon 8 should be part of the routine screening in all patients diagnosed with sporadic MTC in Greece (10).
We describe a family from the United States, in which 21 members have been identified as carriers of the G533C mutation. Eleven additional members of this family are at risk to have inherited this mutation but have not yet been tested. In our family, three members from three different generations have been diagnosed with a pheochromocytoma, the youngest (age 29) being asymptomatic and identified solely by genetic and preoperative screening based on family history.
In 2009, the American Thyroid Association issued specific MTC guidelines in which recommendations regarding the timing of prophylactic thyroidectomy and the extent of surgery are guided by a model that uses genotype–phenotype correlations based on available evidence to stratify mutations into four categories (A–D) of risk (3). Using these guidelines, the G533C mutation is categorized as low risk (Category A), and pheochromocytoma is described as a rare feature associated with this mutation. Accumulating evidence in recent years suggests that this mutation may pose a larger risk than previously thought, as several cases with metastatic disease (both locoregional as well as distant) have been described and pheochromocytoma has been diagnosed in 10 patients with this mutation (including three patients from our kindred), with pheochromocytomas being bilateral in at least two cases. In one reported case, pheochromocytoma was diagnosed eight years after initial detection of this mutation in a large Brazilian kindred (8). Furthermore, the youngest patient to undergo thyroidectomy in this kindred was five years old, and was found to have C-cell hyperplasia, and the youngest with documented MTC and nodal metastases, amongst carriers of this specific mutation reported to date, was 21 years old at the time of diagnosis (4). The youngest member in our kindred with MTC is 22 years old (IV-14). Therefore, we believe that prophylactic thyroidectomy in identified gene carriers under 20 years of age is justified.
Although it is unusual for a diagnosis of pheochromocytoma to precede the diagnosis of MTC in the MEN2A syndrome, three patients in our kindred presented with pheochromocytoma as their initial manifestation; two of them were 29 years old at the time of this diagnosis, and in one of them, a 5 cm pheochromocytoma was diagnosed by preoperative screening in the setting of a known RET gene mutation, despite being normotensive and otherwise asymptomatic (Fig. 2). This patient had a prophylactic thyroidectomy, and histopathology demonstrated bilateral focal CCH; the other one has not yet undergone thyroid evaluation (35 years after resection of pheochromocytoma at the Mayo Clinic in 1977). This patient has three children who have not yet been tested. The third patient with pheochromocytoma is deceased. However, his medical records were reviewed, and we confirmed that he underwent resection of a 10.5 cm pheochromocytoma at a hospital in Illinois. A diagnosis of MTC had not been made. This presentation is similar to that reported previously in two unrelated Greek families (5,7) in whom pheochromocytoma was the initial manifestation of the syndrome before the diagnosis of MTC. Interestingly, our family is also of Greek ancestry, and the possibility of a common link between our family and those previously reported cannot be ruled out.
When recent MTC guidelines were initially published in 2009, only four kindreds with this mutation had been described, and of these, only three patients (from two of these families) had pheochromocytoma. Since then, additional reports (including our own) suggest that pheochromocytoma is not so rare a manifestation in patients with this mutation as initially thought. Furthermore, a careful review of these kindreds and members of our affected family shows that in as many as 18% (22/120 patients; range 5–28%) of cases, metastases (either locoregional or distant) were present (4,6,8), and some patients died of/with metastatic MTC, indicating that this mutation may, in some cases, be associated with a more aggressive phenotype than initially described.
Although a strong genotype–phenotype correlation has been described, clinical heterogeneity is often seen within and among families with the same RET gene mutation, as demonstrated in most described kindreds, including our own. Recently published data suggest that certain polymorphisms (such as the IVS1-126G>T) within the RET proto-oncogene may predispose to early development of MTC in some families with this specific G533C mutation, or influence the age of onset or other clinical manifestations (12). Other clinical features, such as the presence of lymph-node metastases at the time of diagnosis, were more commonly seen in patients with the IVS8 +82A>G polymorphism (12). It is possible, therefore, that the presence or absence of certain polymorphisms (or alterations in unidentified modifier genes) may explain why, in some patients, pheochromocytoma may be the dominant or earliest manifestation of this syndrome, while others present with more advanced features of MTC instead. Additional studies are needed to determine better the specific impact of genetic modifiers, such as the polymorphisms described, as well as environmental modifiers on the clinical presentation in association with the G533C mutation.
Footnotes
Acknowledgment
The authors wish to thank Mrs. Jane Bennett for her assistance in the preparation of this manuscript.
Author Disclosure Statement
No competing financial interests exist.