
Abstract
Background:
Resistance to thyroid hormone (RTH) is a rare condition usually diagnosed in patients with classic thyroid function tests (TFTs) of elevated thyroid hormone levels with nonsuppressed TSH. The presence of autoimmune thyroid disease (AITD) can confound the clinical diagnosis of RTH. A family was evaluated because several members had elevated TSH and normal or low serum T4 concentrations with AITD. While these individuals were initially reported to have RTH, they were found to have a normal thyroid hormone receptor beta (THRB) gene sequence, and three other asymptomatic family members were found to harbor the variant TRβ G339S.
Methods:
The THRB gene was sequenced in 19 members of a large Mexican/Aztec family. In vitro expression of the mutant TRβ protein was performed, as well as computer modeling of the variant compared to known mutations in the flanking codons.
Results:
Investigation of an individual with AITD who was incorrectly diagnosed with RTH led to the fortuitous discovery of a THRB gene variant (G339S) in the proposita's father, paternal aunt, and cousin. This variant was not detected in analysis of 124 unrelated alleles. All individuals harboring G339S had normal TFTs. Normal in vitro expression and function of G339S and molecular modeling predicted that this variant would not have an effect on the hypothalamic–pituitary–thyroid axis as determined by thyroid hormone binding in vitro and thyroid function tests in vivo, despite profound effects seen in mutations in the adjacent codons 338 and 340.
Conclusion:
We report an individual with normal TFTs and AITD harboring a novel THRB gene variant. In addition to illustrating the importance of accurate diagnosis of thyroid disease so that proper treatment and counseling can be given, TRβ codon 339 is not essential for normal TRβ function.
Introduction
F
In all cases of RTH reported to date, identification of the subject was based on the characteristic phenotype of goiter, high serum thyroid hormone levels, and nonsuppressed TSH. We describe a family with autoimmune thyroid disease (AITD) and hypothyroidism in which three family members were incidentally found to harbor a THRB gene variant resulting in the amino acid change G339S. This family was evaluated because several members had elevated TSH and normal or low serum T4 concentrations with AITD. While these individuals were initially reported to have RTH (7), they were now found to have normal THRB gene sequence, and three other asymptomatic family members were found to harbor the mutant TRβ G339S. Of note, mutations in codons flanking the amino acid 339, namely 338 and 340, have been shown to cause the RTH phenotype. This case demonstrates the importance of careful analysis of phenotype and genotype to diagnose TH abnormalities.
Patients and Methods
Case description
In 2008, the proposita, then a 28-year-old Mexican female presented with symptoms of headache, palpitations, fatigue, a 3 kg weight loss in 1 month, hair loss, menstrual irregularities, and insomnia. Physical examination revealed tachycardia and goiter. Pertechnetate-99m scan showed diffuse uptake. Thyroid function tests showed: total T4 11.5 μg/dL (normal: 5–11.6); total T3 188 ng/mL (normal: 80–190); free T3 7.36 pg/mL (normal: 3–7); free T4 1.11 ng/dL (normal: 0.7–1.4); TSH 7.22 μU/mL (normal: 0.4–3.6); and positive thyroperoxidase (TPO) autoantibodies. The family history was remarkable for the patient's mother and two maternal aunts who were being treated for hypothyroidism. The patient was diagnosed with autoimmune hyperthyroidism and was treated with methimazole, despite the nonsuppressed TSH. On this treatment, T4 and T3 normalized and improvement of symptoms were noted. The patient self discontinued the methimazole and again became symptomatically hyperthyroid, although TSH was never suppressed despite increase in both the free T3 or T4. Because of the nonsuppressed TSH, a thyrotropin-secreting adenoma (TSHoma) was suspected and ruled out by a normal magnetic resonance imaging (MRI) and normal response to thyrotropin releasing hormone (TRH) stimulation, that is, there was a normal TSH increase and the α-subunit remained low. The patient was therefore reported as having RTH (7). Blood samples from the proposita and family members, as well as the controls described below, were drawn after obtaining a written consent and were sent to the University of Chicago for further analysis. The protocol has been approved by the Institutional Review Board of the University of Chicago.
Thyroid function tests
Total T4, total T3, and TSH were measured by chemiluminescence immunometric assays using the Elecsys Automated System (Roche Molecular Biochemicals GmbH and Hitachi, Ltd., Indianapolis, IN). Total T3r was measured by RIA (Adaltis Italia S.p.A, Bologna, Italy), and thyroglobulin was measured by an in-house RIA. Serum FT4I and FT3 index were calculated as the product of their serum total concentrations of each iodothyronine and the normalized resin T4 uptake ratio. Antibodies against thyroglobulin (TG) and TPO were measured by passive hemagglutination (Fujirebio, Inc., Tokyo, Japan).
Genetic analysis
Genomic DNA was extracted from whole blood using the commercial QIA amp DNA blood kit (Qiagen, Promega Valencia, CA). Exons 7–10 of the THRB gene were amplified by polymerase chain reaction (PCR) with four sets of primers (Supplementary Table S1; Supplementary Data are available online at
Exons 1–10 of the TSH receptor (TSHR) gene were amplified with 14 sets of primers (Supplementary Table S1). The PCR conditions were as follows; an initial “touchdown” with denaturation for 30 sec (94°C), annealing for 45 sec (63°C), and elongation for 45 sec (72°C) for a total of seven cycles. That was followed by a denaturation for 30 sec (94°C), annealing for 30 sec (57.5°C), elongation for 45 sec (72°C), and finally elongation for 7 min (72°C). All PCR products were electrophoresed on a 1.8% agarose gel.
All PCR samples were sequenced using automated fluorescence-based sequencing (373OXL 96 capillary, Applied Biosystem Carlsbad, CA).
Screening
In order to determine whether the identified THRB gene mutation was a polymorphism in the Mexican population, whole blood was obtained from 62 random, unidentified patients from Hospital Ángeles de Querétaro. Genomic DNA was extracted. Primers were designed such that the mutant allele created a restriction site for the restriction enzyme BsaW1 (New England Biolabs). The primers were as follows. Forward: 5′ GGG AAA TGG CAG TGA ACC GG 3′; reverse: 5′ AGT GTC ATC CAG GTT GAA AGA AG 3′. The PCR conditions were the same as those described for the sequencing of the THRB gene except for the annealing temperature, which was 56.8
Construction of plasmids and cell culture
An expression vector for G339S was constructed by site directed mutagenesis (Quickchange II; Stratagene, La Jolla, CA). The correct placement of the mutation was verified by direct sequencing. A plasmid expressing the firefly luciferase (Luc) driven by three copies of a palindromic TH response element, AGG TCA-TGA CCT, (Palx3-Luc) and positively regulated by T3 was used as reporter (8). Renilla TK was used an internal control.
COS-7 cells were maintained in Dulbecco's modified Eagle's medium (DMEM; Gibco, Grand Island, NY) supplemented with 100 U penicillin and 100 μg of streptomycin per mL and 10% fetal bovine serum (FBS) as previously described (9). On the day of transfection, cells were washed twice with Hank's solution (Gibco). Cells were transfected with Fugene HD (Promega) in a 4:1 ratio. Four to 5 h after the transfection, 500 μL of DMEM+10% of FBS from a thyroidectomized calf+1% penstrep was added. Twelve hours later, 500 μL of the same medium containing 10−7 mol/L T3 or 2×10−10 mol/L T3 were added. The amount of DNA was 1 μg of Palx3-luc, 10 ng of the vector pRL-TK, and 30 ng of WT TRβ, mutant TRβ or empty vector.
After 48 h of incubation, cells were harvested and analyzed for firefly and Renilla luciferase activities (Dual-Luciferase Reporter Assay System; Promega). All experiments were performed in quadruplicate or triplicate for a minimum of three times.
Modeling
Modeling of the G339S substitution was done using the web-based programs Rosetta Backrub (
Results
The proposita (II-1), her sister (II-2), mother (I-2), aunt (I-1), and father (I-3) have TPO and/or TG autoantibodies (Fig. 1). None of the family members had a phenotype consistent with RTH (high T4 and T3 with normal or slightly elevated TSH). Several members had a high TSH (I-1, I-2, I-3, I-4, II-1, II-2, II-10). It should be noted that the mother (I-2) had a markedly elevated TSH on two measurements (85 and 56 mU/L). Despite the inconsistency in the TFTs, THRB and the TSHR genes were sequenced from genomic DNA of the proposita and all members of her family. However, no mutations were identified in the proposita. Coincidently, a point mutation in codon 339 (glycine GGC to serine AGC) in the THRB gene was found in the proposita's father (I-3), who had normal thyroid function tests and was asymptomatic for thyroid disease. The G339S mutation was also found in a paternal aunt (I-6) and in a cousin (II-10) on the father's side of the family. It was not possible to test the individual I-8 (Fig. 1).
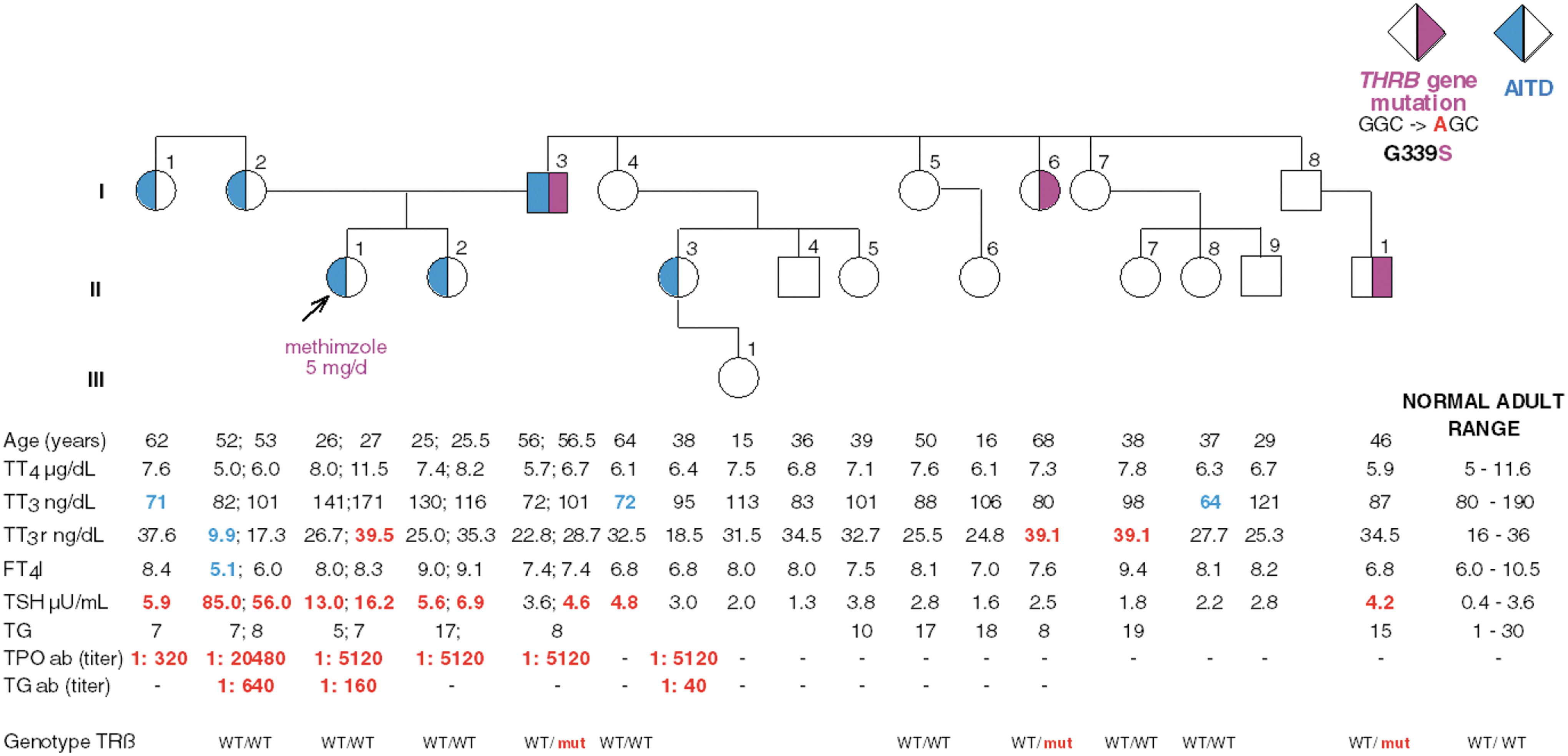
Pedigree and results of thyroid function tests. Square symbols indicate males, circles indicate females, Roman numeral to the left of the pedigree indicates the generation, and numerals on the right of each symbol indicate individual family member. Shaded symbols to the right indicate that a subject have the mutant TRβ G339S (I-3, I-6, II-10). Shaded symbols to the left indicate that a subject is positive for autoantibodies (I-1, I-2, I-3, II-1, II-2, II-3). Subject I-8 was not available for investigation but must also harbor the mutation because his son II-10 has the mutation. Abnormal values above the upper limit of normal are in red and abnormal values below the lower limit of normal are in blue. The arrow indicates the proposita. AITD, autoimmune thyroid disease; T3, 3,3′,5-triiodothyronine; T3r, 3,3′,5′-triiodothyronine; T4, thyroxine; TT3, TT3r, and TT4, total T3, T3r, and T4, respectively; FT4I, free T4 index; TSH, thyrotropin; TG, thyroglobulin; TPO, thyroperoxidase; ab, autoantibodies; WT, wild type.
To exclude the possibility that G339S was a polymorphism in this population, we obtained blood from 62 random unrelated patients from the same hospital where the proposita has been treated. A total of 124 alleles were screened, and none had the G339S mutation.
Although the G339S has not been described before, several mutations have been reported in the two flanking codons, 338 and 340, in subjects with typical RTH phenotypes (Table 1). R338W, R338L, G340R, and Q340H have high free T4 or high free T4I and normal TSH consistent with RTH.
TSH, thyrotropin; T4, thyroxine.
It was important to investigate the expected structural consequences of the mutation. The lack of side chain on Gly permits a larger range of backbone values in determining the course of the protein chain segment containing it. Additionally, its nearest neighbors might be affected by addition of even the small side chain of Ser. We used the web-based program RosettaBackrub (10), the point mutation application, to model the G339S substitution into the high-resolution crystal structure of TRβ ligand-binding domain (PDB 3GWS). The program generated 20 conformations of the affected region, within the 6 Å distance from the position 339, sampled based on the probability scores. Modeling showed (Fig. 2, upper inset) that a G to S substitution in this position should leave the 3D structure unchanged. Specifically, residue 339 is located on the first turn of a short α-helix (H7). With its side chain facing into solvent Serine in this position would not affect the secondary structure; an α-helix will remain an α-helix, and possible bending of the helix would not be noticeable because this helix is too short. Unlike the flanking residues R338 and Q340, G339 is not involved in forming network interactions with neighboring residues (Fig. 2, lower inset). The substitution G339S is unlikely to change network interactions, and therefore the structural impact of the region is small. Residue 339 is solvent exposed, positioned on the surface distant from the potential interfaces involved in TRβ activation and regulation such as hormone binding pocket, helix H12 [activation function 2 (AF2) site], and potential dimerization interface (helices H10 and H11; Fig. 2). The impact of G339S substitution on thermostability of the TRβ ligand-binding domain was also evaluated using the programs CUPSAT (11) and I-Mutant 2.0 (12). The predicted effects (ΔΔG) were very minor, 1.02 kcal/mol (CUPSAT) and −0.23 kcal/mol (I-Mutant 2.0), indicating that the thermostability of the mutant is comparable to the wild type protein. Therefore, modeling predicts that biological consequences of mutation G339S seem unlikely. We note that hydroxyl group of the Ser, which is certain to be solvated by associated water in vivo, might be able to form interactions with protein binding partners that might interact at this site. However, none are known.
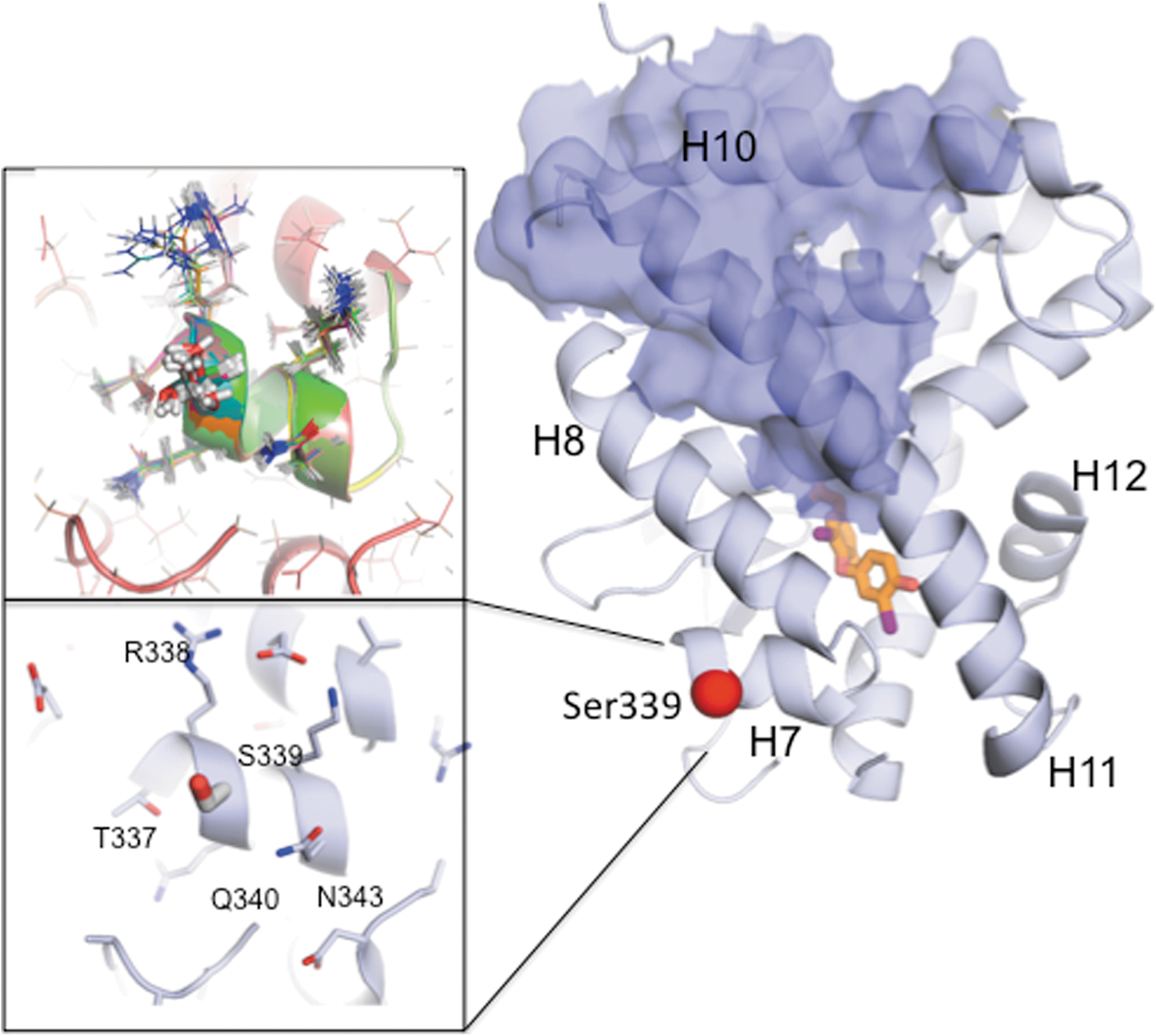
Modeling of the G339S mutation on the structure of TRβ. Modeling has been done using atomic coordinates of ligand binding domain of human TRβ bound to T3 (3GWS.pdb). The structure of TRβ is shown as a cartoon; some helices are indicated. The hormone is shown as a stick model with carbons in orange, iodines in magenta, oxygens in red, and nitrogens in blue. The surface presumably involved in homodimerization and heterodimerization with RXR is shown in blue. The position of mutation G339S is indicated using the red sphere. Lower inset: The area surrounding position 339 with Ser modeled in is shown in detail, and the neighboring residues are indicated. Upper inset: The superposition of 20 predicted conformations of the same area calculated using Rosetta Backrub is shown. The sidechains are shown as stick models with modeled Ser 339 being show as a thick stick model.
In vitro studies showed that the stimulatory response to T3 was no different with the 339S and the WT TRβ at any T3 dose. In the presence of 2×10−10 mol/L T3, the relative Luc activity of the WT TRβ and 339S using PALx3 was 846±142 and 1061±199 respectively. At the higher dose of 10−7 mol/L T3, no significant difference was observed. The relative Luc activity of the WT TRβ and 339S was 1746±153 and 1262±823. In contrast, significant reduction in the response was observed in the 338W mutant at both low and high T3 doses of 402±159 and 657±272 respectively (Fig. 3).
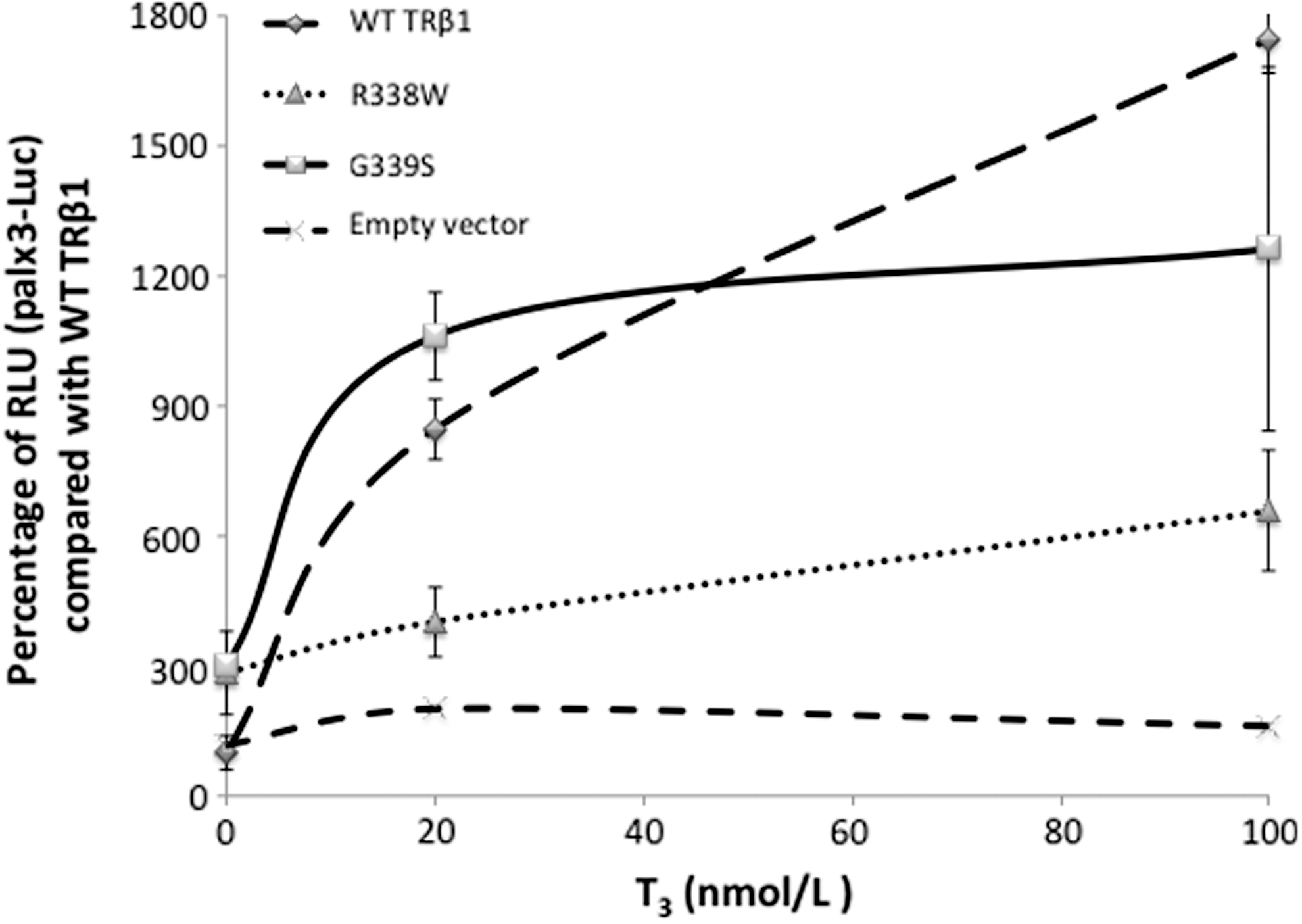
In vitro functional analysis of TRβ1. Analysis of 339S, and 338W compared with WT G339 and R338. Cos-7 cells were transfected with the empty vector to determine the background luciferase activity. Results are expressed relative to those obtained with the WT TRβ1. Data shown are from a single experiment. In contrast to the mutant 338W, the mutant 339S does not show a reduced stimulation with T3 compared to the WT. Error bars represent standard error of the mean. RLU, relative luminescence units.
Discussion
We present an incidental finding of a THRB gene variant that is without effect on thyroid function tests in a family with AITD. Fortuitously, the proposita was misdiagnosed with RTH, and samples from the family were analyzed, thus leading to the identification of the G339S in the asymptomatic father of the proposita. We cannot exclude effects on other tissues, although in vitro data indicate that the variant TRβ is unlikely to have any effect. This report illustrates the problems with diagnosis of abnormal thyroid function when a combination of TFTs are not considered. In the case of the proposita, there was no set of TFTs that had both elevations of free T3 and free T4 with nonsuppressed TSH. In addition, the presence of thyroid autoantibodies should prompt the physician to do an additional evaluation evaluating for interference. We excluded nonTR-RTH because she did not have the biochemical phenotype or clinical symptoms such as goiter. Serum analysis in the Chicago laboratory demonstrated high TSH and normal free T4 and free T3 values. This could also have been a result of the methimazole treatment or resistance to TSH (RTSH) (13). Patients with RTSH usually present with hyperthyrotropinemia and sometimes low T4, T3, and symptoms of hypothyroidism, although the proposita reported herein was symptomatically hyperthyroid. However, the sequence of the TSHR gene in the proposita was normal. Thus, we concluded that the symptoms of the proposita are due to AITD and not RTH. The other family members with positive thyroid autoantibodies (I-1, I-2, I-3, II-2) have mild hypothyroidism (or “subclinical hypothyroidism”) based on their TFTs. The clustering of the phenotype in this kindred could suggest a genetic disease, AITD being one, even though not monogenic (14). It is important to distinguish between AITD and RTH because the treatment is different, especially during pregnancy. In AITD, it is important to control TH, but in RTH, the mother compensates with elevated TH and should not be normalized if the fetus is similarly affected (15).
While codons flanking G339 do result in a marked phenotype of RTH, the mutation G339S does not. This may be due to unique properties of the amino acid substitution at this position, which could be neutral in conferring structural change. The father (I-3) who harbors the G339S variant has normal TFTs except for a slightly elevated TSH, but he also has positive TPO antibodies. The two other subjects (I-6, II-10) who harbor the same variant also have normal TFTs except for II-10 who also has a slightly elevated TSH. Structural modeling of G339S did not predict loss of function, which was confirmed by in vitro studies. It should be noted that the in vitro studies are limited by the fact that we only used a palindromic positive response element with only two doses of T3. Receptor function might be different on different types of positive or negative elements and if lower doses of T3 were used. There are no studies to suggest that the clinical symptoms of RTH worsen with age. Thus, with no symptoms and normal TFTs, we do not recommend further follow-up for the individuals harboring this mutation. However, it is known that the symptoms vary from patient to patient (16). Catargi et al. (17) presented a family in which the proposita was asymptomatic her whole life, whereas the mother and grandmother had a classic RTH phenotype. However, in the current case, knowledge that the variant does not result in impaired binding indicates that no further testing is necessary.
The coexistence of AITD and RTH caused by THRB gene mutations has been previously studied (18), and it has been shown that the presence of a mutant TRβ is associated with an increased incidence of AITD. However, in the cohort of patients examined in that study, all subjects were symptomatic and had clearly abnormal TFTs. The mechanism of the association remains unknown. The findings presented here illustrate the importance of accurate diagnosis of thyroid disease so that proper treatment and counseling can be given.
Footnotes
Acknowledgments
This work was supported in part by Grants DK15070, DK091016, and RR04999 from the National Institutes of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Diabetes and Digestive and Kidney Diseases or the National Institutes of Health.
Author Disclosure Statement
No competing financial interests exist.