
Abstract
Background:
Thyroid dysfunction is common in newborn infants with Down's syndrome (DS), but defects causing classic thyroid dysgenesis (TD) with permanent congenital hypothyroidism (CH) have not been described.
Objective:
We studied a girl with DS and CH who had a mutation in the promoter sequence of the PAX8 gene.
Results:
A female infant was found to have trisomy 21 and CH, with a venous thyrotropin (TSH) of >150 mU/L and a free thyroxine (fT4) of 15.1 pmol/L (day 12). Thyroid peroxidase antibodies and thyroglobulin antibodies were elevated. Scintigraphy showed normal uptake, but ultrasound identified a small gland with heterogenous echotexture and cystic changes. Sequence analysis of the PAX8 gene revealed a new heterozygous maternally inherited mutation (−3C>T) close to the transcription initiation site. Electromobility shift assay studies of the wild type and the mutant PAX8 sequence incubated with nuclear extracts from PCCL3 cells exhibited that the sequence at position −3 is not involved in specific protein binding. However, the mutant PAX8 promoter showed a significantly reduced transcriptional activation of a luciferase reporter gene in vitro tested in HEK, PCCL3, as well as in HeLa cells, indicating that this mutation is very likely to lead to reduced PAX8 expression.
Conclusions:
The persistent CH in this patient with DS is likely to be attributable to the diminished PAX8 expression due to a new heterozygous mutation in the PAX8 promoter sequence. Our case shows that true CH may occur in DS, as in the general population. Furthermore, it is possible that the trisomy 21 itself may have resulted in a more severe phenotypic expression of the PAX8 mutation in the child than the mother.
Introduction
C
Down's syndrome (DS) is the most common chromosomal disorder with an adjusted prevalence in the United Kingdom of 1.08 per 1000 live births (7). Thyroid dysfunction is common, particularly acquired autoimmune thyroiditis (8). In newborn infants, thyrotropin (TSH) elevation is often seen and is typically transient in nature (9), possibly reflecting altered regulation of the thyroid axis (10). However, classic CH with dysgenesis is not an established feature in DS.
We describe a new mutation close to the transcription initiation site of the PAX8 gene in a girl with DS and thyroid hypoplasia. In vitro studies show that this mutation causes a reduction in the PAX8 gene expression level, and is therefore very likely to be causative.
Case Report
Our patient, a female, was born after 40 weeks of gestation by spontaneous vaginal delivery as a breech presentation to a 24-year-old mother with no history of thyroid disease following a normal pregnancy. Dysmorphic features consistent with DS were noted shortly after birth, and karyotype examination confirmed trisomy 21. A cardiac ultrasound showed a large perimembranous ventricular septal defect (VSD), a secundum atrial septal defect (ASD), and a patent ductus arteriosus (PDA). Elective repair was planned, but at 35 days, the baby was admitted with failure to thrive and cardiac failure. She was commenced on nasogastric feeding and regular diuretics, and subsequently underwent Dacron patch closure of her VSD, a direct suture closure of her ASD, and a ligation of her PDA at the age of 69 days. She was readmitted on day 106 with respiratory distress and stridor, and was found to have supraglottic edema consistent with reflux laryngitis. A barium swallow confirmed significant gastroesophageal reflux, and she underwent fundoplication and gastrostomy insertion at day 133. Her gastrostomy has remained in situ since discharge, and she required readmission at the age of 2.25 years for open jejunostomy and reinsertion of the gastrostomy.
Thyroid features
Newborn blood spot screening on day 5 demonstrated hyperthyrotropinemia at 43.8 mU/L in whole blood (normal<8 mU/L). At the time of referral (day 12), she was noted to be constipated and suffering from prolonged neonatal jaundice, and a venous blood sample showed a serum TSH of >150 mU/L (normal 0.72–13.1 mU/L) and a normal free thyroxine (fT4) of 15.1 pmol/L (normal 8–25 pmol/L) indicating compensated primary CH. Thyroid peroxidase antibodies (TPOAb) were elevated at 409 IU/mL (normal <50 IU/mL) consistent with transplacental transfer of maternal thyroid antibodies. Unfortunately, however, there is no record of maternal thyroid function tests from this time. The infant was commenced on levothyroxine at 25 and 50 μg on alternate days. Thyroid function tests were repeated on day 28 and demonstrated a good response to levothyroxine with a serum TSH of 11.03 mU/L (normal 0.72–13.1 mU/L), a fT4 of 31.9 pmol/L (normal 8–25 pmol/L), and a free triiodothyronine (fT3) of 2.2 nmol/L (normal 0.9–2.8 nmol/L). Her thyroglobulin was 103 ng/mL (normal <55 ng/mL), and thyroglobulin antibodies were<20 IU/L (normal <40 IU/L). In light of these results, her levothyroxine dose was adjusted to 25 μg daily.
Combined ultrasound and radioisotope imaging of the thyroid gland was performed on day 14. An ultrasound showed a small gland with a right lobe volume of 0.44 mL, a left lobe volume of 0.31 mL, and a combined volume of 0.76 mL (reference range 1.0–3.3 mL) (6). The gland was very heterogeneous in echotexture with many small and two large cystic areas. The isthmus appeared bulky, measuring 0.26 mL. A radioisotope thyroid scan after intravenous injection of 10 MBq of pertechnetate showed a normal uptake in an apparently normal bilobed thyroid gland in a normal position.
At 1.35 years, her TSH was very elevated at >150 mU/L (normal 0.46–8 mU/L) with a fT4 of 9.3 pmol/L (normal 9–23 pmol/L) despite treatment with 43.75 μg of levothyroxine daily, suggestive of compliance difficulties. The levothyroxine dose was increased to 50 μg daily, and her thyroid function remained normal thereafter.
At 3.8 years, the dose of levothyroxine was reduced to 25 μg daily to establish whether the child's CH was transient or permanent in nature. Six weeks later, her TSH had risen to 53.61 mU/L (reference range 0.35–5.5 mU/L) with a fT4 of 12.1 pmol/L (reference range 9–24 pmol/L) confirming the persistence of primary hypothyroidism, and the levothyroxine dose was increased back to 50 μg daily.
At the same time, the mother was found to have a TSH of 16.13 mU/L, a fT4 of 12.6 pmol/L, and TPOAb >1000 IU/mL, and treatment with levothyroxine was initiated.
After obtaining informed written consent, molecular genetic studies were conducted.
Material and Methods
Subjects, DNA amplification, and DNA sequencing
We obtained written informed consent from all available family members to participate in the clinical and genetic studies. Genomic DNA from blood samples was isolated utilizing the Puregene® Blood Core Kit B (Qiagen, Hilden, Germany). The coding sequences of the candidate genes for TD, as well as the promoter sequence of the PAX8 gene, were amplified by polymerase chain reaction (PCR; conditions and primer sequences for mutational screening of TSHR, PAX8, TTF1, TTF2, and NKX2.5 can be supplied upon request), purified with the HiYield® PCR Clean-up/Gel Extraction Kit (Süd-Laborbedarf GmbH, Gauting, Germany), and then sequenced with BigDye terminator v.3.1 purified by ethanol precipitation and analyzed using an automated sequencing system (3130 Avant Genetic Analyzer, Applied Biosystems, Weiterstadt, Germany).
Constructs and mutagenesis
To study the consequence of the PAX8 promoter mutation, a 1130 bp fragment upstream of the ATG of the PAX8 gene was amplified and cloned upstream of the luciferase gene of the pGL3 basic vector (Promega, Madison, WI). This promoter construct (PAX8proWT) was a gift from Dr. Peter Kopp (Northwestern University, Chicago, IL). To construct the PAX8 promoter mutation (PAX8proMUT), we used the Quick Change Mutagenesis XL kit (Agilent Technologies, La Jolla, CA) and the primers PAX8_-3-F: 5′-GGT GAT GCC GGG TGA ATG GGA ACA AAC-3′ and PAX8_-3_R: 5′-GTT TGT TCC CAT TCA CCC GGC ATC ACC-3′ respectively. All constructs were verified by sequencing.
Cell culture and transfection
HeLa and HEK cells were grown in PAN 401 medium supplemented with 10% fetal bovine serum (FBS; PAN Biotech GmbH, Aidenbach, Germany) and 1% penicillin-streptomycin (PAN Biotech GmbH) at 37°C in a humidified chamber with 5% CO2. PCCL3 cells were a gift from Dr. Annette Altmann (Clinical Cooperation Unit, Nuclear Medicine, Heidelberg, Germany), and were grown in Ham's F12 K medium supplemented with 10% FBS, 1% penicillin-streptomycin (PAN Biotech GmbH), 10 ng/mL somatostatin, 10 ng/mL Glys-His-Lys, 5 μg/mL transferin, 10 nM hydrocortisone, 10 μg/mL insulin, and 10 mU/mL TSH (Sigma-Aldrich, St. Louis, MO) at 37°C in a humidified chamber with 5% CO2. Twelve hours before transfection, cells were trypsinized and plated in wells on a 24-well plate. When the cells reached a confluence of 70%, transfection with FuGene HD (Roche, Mannheim, Germany) was carried out according to the manufacturer's instructions. The ratio of the transfection reagent to DNA was 2:3. Forty-eight hours after transfection, the cells were washed three times with ice cold phosphate-buffered saline (PBS; Invitrogen, Carlsbad, CA) and lysed in 150 μL of 1× passive lysis solution (Promega). Ten μL of the protein extract was used for the dual luciferase assay (Promega). The measurement of the firefly and the Renilla luciferase activity was performed according to the manufacturer's recommendations.
Electromobility shift assay (EMSA)
The proteins from nuclear extracts from HeLa, PCCL3, and HEK cells were isolated as described previously (11). The wild type as well as the mutant PAX8 promoter oligo sequences were 5′-end-labeled with IRD700 (Metabion, Martinsried, Germany). The 10 μL binding reaction contained 1 μg of protein, 10–50 nM oligo, 150 ng poly (dI-dC), 500 ng salmon sperm DNA, 5 mM DTT, 0.5% Tween20, 10 mM Tris, and 50 mM NaCl pH 7.5. The binding reaction was incubated for 30 min at room temperature. Afterwards, 1 μL gel-loading buffer (containing 650 mg/mL sucrose, 0.3% OrangeG, 10 μM Tris pH 7.5, and 10 μM EDTA pH 8.0) was added, and the samples were loaded on a 4% native polyacrylamide (PAA) gel. The PAA gel was supplemented with 50 mM Tris pH 7.5, 200 mM glycine, 2 mM EDTA. Towbin buffer (12) supplemented with 10 mM EDTA served as a running buffer. Thereafter, the PAA gels were scanned and visualized using the Odyssey Model 9120 and the Odyssey v2.1 software (Li-Cor Biosciences, Bad Homburg, Germany).
To test the protein binding capacity of the wild type and the mutant promoter, the primers used for mutagenesis of PAX8proWT and PAX8proMUT were labelled with an infrared dye (IRD700) and incubated with proteins of a nuclear extract isolated from HeLa cells, PCCL3 cells, or HEK cells.
Results
Sequence analysis of the proposita's genomic DNA revealed a heterozygous mutation in the promoter sequence of the PAX8 gene at position −3 upstream of exon 1 (G>A). This is 562 bp upstream of the translation start site in exon 2. This mutation is located in a highly conserved area of the PAX8 gene promoter (data not shown). It was not found in 100 normal alleles, making a common polymorphism unlikely. The mutation was also found in a heterozygous state in the mother of the proposita.
To study the consequence of the nucleotide substitution in the PAX8 promoter region, both reporter constructs (PAX8proWT and PAX8proMUT) driving a luciferase reporter gene were transiently transfected into HeLa, PCCL3, and HEK cells respectively. Using 1 μg of plasmid DNA of each promoter construct, a significant decrease of the luciferase expression was observed with the PAX8proMut construct compared to the PAX8proWT in all three cell lines (Fig. 1). We then investigated the ability of nuclear extracts from HeLa, HEK, and PCCL3 cells to bind to the investigated PAX8 promoter sequence. Neither the investigated wild type promoter sequence nor the mutant PAX8 promoter sequence showed a specific shift (Fig. 2), indicating that this sequence is not important for specific protein binding.
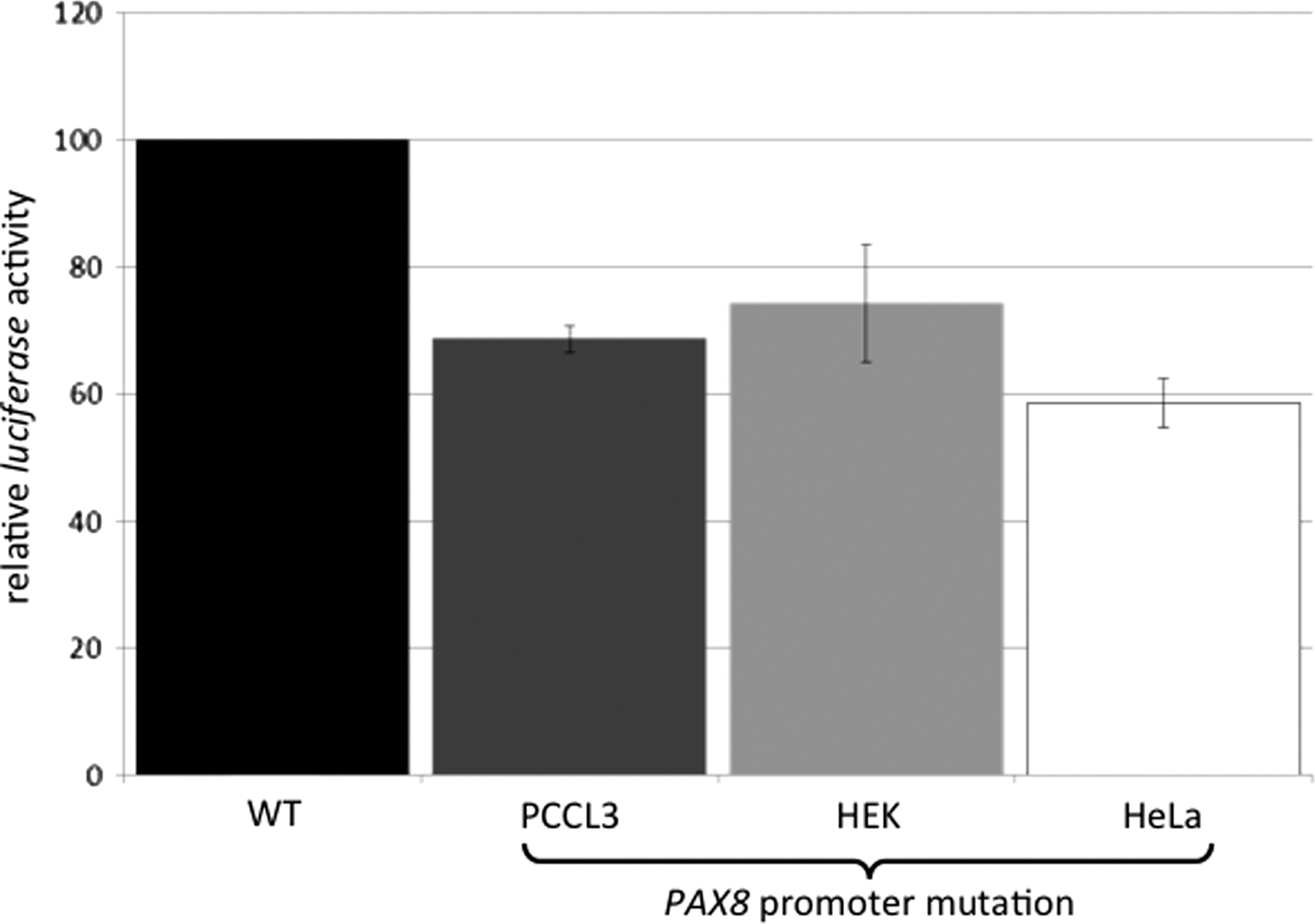
In vitro effects of the PAX8 promoter mutation. Both PAX8 gene promoter constructs (PAX8proWT and PAX8proMUT) were transiently transfected into PCCL3, HEK, or HeLA cells. Using 1 μg of plasmid DNA of each promoter construct, a significant decrease of the luciferase expression was observed with the PAX8proMut compared to the PAX8proWT (dark black bar) in all three cell lines.

Electromobility shift assay (EMSA) with nuclear extracts from PCCL3 and PAX8proWT and PAX8proMUT. Nuclear extracts isolated from PCCL3 cells were incubated with double stranded (ds) oligonucleotides containing either the wild type or the mutant PAX8 promoter sequence. No specific protein binding to either double stranded oligo was observed.
Discussion
We report the case of a girl with DS who had CH attributable to thyroid hypoplasia associated with a heterozygous mutation close to the transcription initiation site sequence of the PAX8 promoter, which was inherited from the mother. In our case, there was evidence of maternal Hashimoto's disease in that elevated TPO antibodies were detected in the infant during the postnatal period, while the mother showed very high TPO antibodies together with compensated primary hypothyroidism (normal fT4 but elevated TSH) when the girl was reevaluated at 3.5 years of age.
It is known that infants with DS commonly show TSH elevations (9,10), which raises the possibility that the thyroid dysfunction in our case could simply represent a nonspecific effect of DS. Alternatively, it could be argued that the CH in our patient resulted from transplacental transfer of maternal thyroid antibodies.
Concerning the first point, it is true that TSH elevations occur more commonly in DS, as for example when detected by newborn screening, compared with the general population (13,14). However, there is no evidence that the incidence of the classic cause of true permanent CH—TD—is more common in DS. Indeed, studies to date have shown the thyroid gland is neither ectopic nor absent in DS. Fort et al. described the clinical characteristics of 11 DS newborns detected by neonatal screening, none of whom had athyreosis or thyroid ectopia (13). Moreover, a recent study of 13 fetuses with DS by Luton et al. found no abnormality, including goiter, on ultrasonography, although heterogeneity of the glands and reduced follicular size on histology was found (15). However, it should be noted that detailed, high-resolution imaging of the thyroid in DS has not been carried out in a prospective fashion, and hence mild alterations in size may have previously gone undetected.
By contrast, it is well established that TSH levels tend to be elevated in newborns with DS (9,10). Van Trotsenburg et al. found a shift toward relatively higher TSH and lower fT4 levels in 97 placebo-treated infants with DS, while 99 DS infants treated with levothyroxine needed relatively high fT4 concentrations to normalize TSH (16). This group concluded that a type of mild, persistent CH occurs in DS, possibly related to the genomic dosage imbalance of dosage-sensitive genes interfering with thyroid hormone production. This conjecture is supported by a recent study by Myerovitch et al., who found a significant shift of the TSH distribution curve to higher values compared with the general population (17), and also of Luton et al., who found TSH elevations with low fT4 levels yet no goiter in 6 of the 13 fetuses with DS studied, indicating a degree of TSH resistance (15). Thus, according to van Trotsenburg et al., many infants with DS who are detected with raised TSH in the newborn period may simply represent those at the extreme end of abnormal thyroid function parameters due to what can be regarded as a DS-specific type of CH (16).
It is clear that our case is different from the model of TSH resistance described above. The finding of actual thyroid gland hypoplasia, with dysplastic features comprising cystic changes on ultrasound, is not consistent with the thyroid dysfunction associated with DS in the newborn period reported previously. Similarly, transplacental transfer of maternal antibodies, while known to cause transient CH (18), would not be expected to cause true, permanent CH as in our case, in whom a significant TSH elevation (>50 mU/L) occurred when the levothyroxine dose was reduced from 50 to 25 μg at three years of age.
It is our belief that the CH in our patient with DS is due to the finding of a mutation in the PAX8 promoter. So far, two monoallelic mutations in the PAX8 promoter region have been described in individuals with TD (2,6), but in vitro functional studies were only performed in one case (6). We therefore decided to investigate whether the mutation at position −3 found in our patient is pathogenic by performing in vitro studies. As expected, the mutant PAX8 gene promoter showed a significantly (25%) reduced ability to activate a reporter gene compared to the wild type (Fig. 1). This result is likely to be due to the fact that the mutant has impact on the initiation of the transcriptional process (19). This is because this part of the promoter sequence is recognized by the transcription initiation complex and is thus important for proper binding of the DNA-dependent RNA polymerase II (20). We therefore postulate that −3G> A is very likely to be pathogenic, since this mutation falls directly into the conserved sequence of the transcription initiator element (21).
The fact that both the normal PAX8 promoter and the mutant sequence did not bind specific nuclear proteins present in PCCL3, HeLa, or HEK cells in our EMSA experiments (Fig. 2) is not contradictory to our functional studies of the promoter. This is because the double stranded oligonucleotide investigated covers only a small part of the promoter region. It is possible, therefore, that if a larger piece of the transcriptional machinery had been investigated, the altered sequence might have had an impact on the binding of the transcriptional machinery in contrast to the normal PAX8 promoter. We do not believe that the reduction in transcriptional activity, which we have found, is caused by a thyroid specific factor, since we have observed this not only in thyroid cells (PCCL3), but also in HEK and HeLa cells. Work by Nitsch et al. supports our hypothesis, since this group identified a thyroid-specific transcriptional enhancer element 84 kb upstream of the PAX8 transcription start site (22). Taken together, our in vitro findings suggest that the altered promoter leads to reduced PAX8 gene expression, which is likely to be involved in the pathogenesis of TD in our index patient.
The question arises as to why true permanent CH has occurred in our index case with DS whereas the mother was initially euthyroid, although she developed hypothyroidism due to her Hashimoto's disease. It is known that PAX8 mutations produce a highly variable phenotype (3,4,6,23 –25). It is therefore not surprising that the mother, who is also a heterozygous carrier of the monoallelic PAX8 gene mutation, was initially phenotypically normal. Our case is similar to that of Congdon et al., who reported a girl with CH in association with a eutopic hypoplastic thyroid gland, who had a heterozygous transversion 119A–>C in exon 3 of PAX8 replacing a conserved glutamine by proline in the paired box domain (Q40P) (26). The child's mother was also heterozygous for this mutation but had a thyroid gland of normal size and mild, adult-onset autoimmune hypothyroidism. It is of interest that the mothers in both our study and the report by Congdon et al. had autoimmune hypothyroidism, raising the possibility that a PAX8 alteration might render the individual more prone to autoimmune thyroid disease.
It remains unclear in what way this phenotypic variability is due to variable penetrance or expressivity of the mutant gene, or whether the genetic background plays a role in this context. Moreover, haploinsufficiency, monoallelic expression, or imprinting could impact on the phenotype (5).
It is possible that the CH related to a PAX8 mutation that has been inherited from the mother was completely independent of the child's trisomy 21 in our case. If this is so, our case serves to illustrate that CH, which has an incidence of about 1 in 3000 births, may occur in DS just as it does in the general population, and that infants with DS and significant TSH elevations are entitled to the same investigation, including thyroid imaging, as other infants who are detected on newborn screening. However, it is also possible that the genomic imbalance related to trisomy 21 in our patient may have resulted in a more severe expression of the PAX8 gene mutation, which she had inherited from her mother.
Footnotes
Acknowledgments
We thank Michaela Bartusel and Farah Izadpanah for excellent technical assistance. P.H. was supported from the MAIFOR program, Germany. Furthermore, we thank all family members for their participation in this study, and Dr. Peter Kopp (Northwestern University, Chicago, IL) for providing the PAX8 wild type promoter construct.
Author Disclosure Statement
The authors have nothing to disclose.