
Abstract
Background:
The established paradigm for thyroglobulin (Tg) function is that of a high molecular weight precursor of the much smaller thyroid hormones, triiodothyronine (T3) and thyroxine (T4). However, speculation regarding the cause of the functional and morphologic heterogeneity of the follicles that make up the thyroid gland has given rise to the proposition that Tg is not only a precursor of thyroid hormones, but that it also functions as an important signal molecule in regulating thyroid hormone biosynthesis.
Summary:
Evidence supporting this alternative paradigm of Tg function, including the up- or downregulation by colloidal Tg of the transcription of Tg, iodide transporters, and enzymes employed in Tg iodination, and also the effects of Tg on the proliferation of thyroid and nonthyroid cells, is examined in the present review. Also discussed in detail are potential mechanisms of Tg signaling in follicular cells.
Conclusions:
Finally, we propose a mechanism, based on experimental observations of Tg effects on thyroid cell behavior, that could account for the phenomenon of follicular heterogeneity as a highly regulated cycle of increasing and decreasing colloidal Tg concentration that functions to optimize thyroid hormone production through the transcriptional activation or suppression of specific genes.
Introduction
T
Review
Current/historical view of counter-regulation of TH synthesis
The “classic” view of the regulation of TH synthesis is that of a “negative feedback system” involving reciprocal interactions between the thyroid gland and higher levels of control using information conveyed by circulating hormones. In this system, referred to as the hypothalamic-pituitary-thyroid axis (HPT), the synthesis of TH, that is, triiodothyronine (T3) and thyroxine (T4), in the thyroid is induced by thyrotropin (TSH) secreted from anterior pituitary thyrotrophs. Increased TH, in turn, suppresses further TSH secretion, primarily in an indirect manner by acting on the hypothalamus to inhibit the release of thyrotropin-releasing hormone (TRH). Although often presented as a seemingly simple and straightforward process involving three organs and three cell types, the detailed features of the HPT actually involve complex local interactions between cell types that are now recognized as being critical to the maintenance of TH homeostasis. For example, a recent study by Fonseca et al. highlights the concept that TH inhibition of TRH release involves not only the TRH-secreting neurons, but also the supporting cells that perform the required conversion of T4 to T3, the active hormone in the suppression of TRH synthesis (5). Furthermore, this group showed that the “indirect” action of TH on TRH secretion is mediated by T4 deiodination in the tanycyte (a type of ependymal cell) rather than the astrocyte. Other aspects of the HPT, such as the stimulation of TH biosynthesis by TSH, are also more complicated than is depicted in the simple endocrine feedback loop. For example, TH synthesis requires the presence not only of TSH but also of insulin and insulin-like growth factor-1 acting as essential cofactors in hormonogenesis (6 –10). Some of these factors, including growth factors and other peptides, are released from the thyrocytes and act locally to either stimulate or inhibit various thyroid functions.
As is currently understood, only one of these factors participates in what could be termed an intrathyroidal negative feedback system. In this process, known as the Wolff–Chaikoff effect, iodide (I−) in excess concentration suppresses its own oxidation and coupling to Tg (organification) and subsequently the production of additional TH (11,12). Inhibition of thyroid function lasts only about two days, however (13), since thyroid cells eventually adapt their iodide transport systems to achieve lower cellular iodide concentrations and escape from Wolff–Chaikoff inhibition (14). The escape from the Wolff–Chaikoff effect includes a reduction of iodide uptake that is associated with a decreased expression of the sodium iodide symporter (NIS, Slc5a5) (15).
The problem of follicular heterogeneity
The thyroid follicle: the morphologic unit of thyroid function
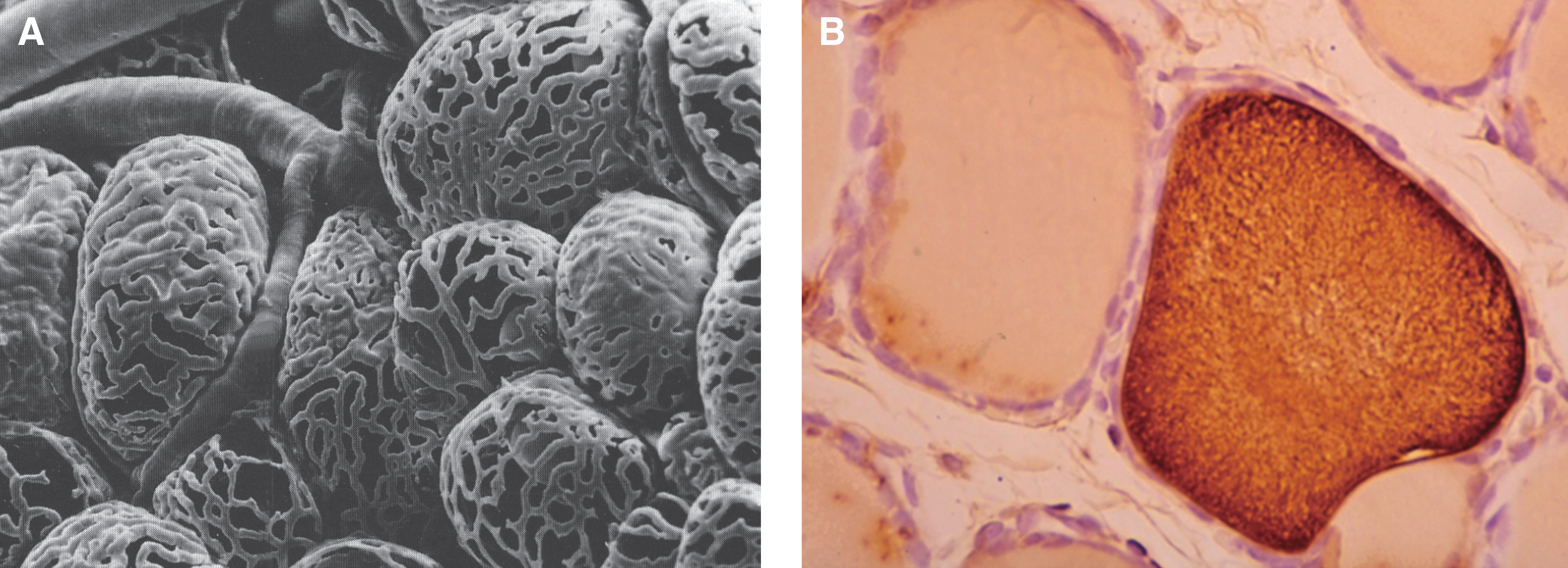
The unique microvascular architecture in the thyroid, with basket-like capillary networks organized around each follicle (Fig. 1A) (16), supports the concept that the follicle, rather than a lobe or a lobule as in many other endocrine organs, constitutes the basic functional unit of the thyroid. This extensive capillary network allows the close contact of the capillary wall and the basal aspect of each thyroid cell to permit the uniform access of all cells in a single follicle to regulatory factors present in the circulation. Each follicle, in turn, receives essentially the same blood supply as any other, allowing circulating TSH to reach the follicular cells in equal concentration throughout the gland and interact with TSH receptors (TSHR) on the basolateral membranes of these cells to trigger intracellular pathways leading to TH biosynthesis. In the normally functioning thyroid at least, the homogeneous distribution of TSHR throughout the gland presents little opportunity for a follicle-specific action of TSH (17). Therefore, with both TSH and its receptor being modulated at the whole organ level rather than at the level of the follicle, it becomes difficult to explain satisfactorily the rather marked differences in follicular form and function throughout the thyroid that we refer to as follicular heterogeneity solely with the existing paradigms of thyroid regulation (18).
Morphological and functional heterogeneity of follicles
It is apparent from even the lowest magnifications of thyroid glands in histologic section that regulation of thyroid function is more complicated than can be accounted for by simple systemic control mechanisms such as fluctuating levels of TSH and iodide. Rather than showing a uniformity of height and width throughout all follicles, the thyroid follicular cells in a normal thyroid gland vary tremendously in dimension and other measures of cell activity from follicle to follicle (19,20). In fact, “active” follicles with tall columnar epithelium are often in direct contact with “inactive” follicles whose cells are in a low cuboidal to almost squamous configuration (21,22). Moreover, other factors such as Tg content and colloid density of individual follicles, and follicular size (allowing for apparent differences due to plane of section) also vary just as greatly. Figure 1B shows a section of rat thyroid gland stained for Tg, in which two adjacent follicles vary significantly in staining intensity. Thus, despite the uniformity of TSH supply to both follicles, their functional states are not synchronized, but rather appear to vary independently of each other.
Follicles are also quite heterogeneous in their distribution of iodide, whose cellular uptake and subsequent diffusion throughout the follicular lumen in covalent association with Tg have been shown to vary widely (23 –25). Heterogeneity also encompasses the differential expression of specific proteins and transcription factors (26 –28) and the differential rates of cell growth from one follicle to the next (29,30). The signaling pathways (cAMP dependent vs. cAMP independent) responsible for regulating thyroid growth also vary from follicle to follicle (31).
It has been argued that follicular heterogeneity could have a genetic or developmental origin, in that individual thyrocytes in primary culture and even cloned thyroid cells vary markedly in functional characteristics (32). In fact, functional heterogeneity of individual thyroid cells is apparent in the earliest stages of thyroid gland development, even before follicles have formed (33,34). As shown in Figure 2A, the rat thyroid anlage adjacent to the developing trachea at embryonic day 15 (E15) is composed of compact rosettes of epithelioid cells apparently devoid of follicles (Fig. 2B). Within this solid anlage, small groups of Tg-positive cells are already identifiable (Fig. 2C, arrows) and are juxtaposed with cells not yet expressing Tg (34). Hence, thyroid cellular function seems to be heterogeneous at the beginning of thyroid development. Despite the early heterogeneity, however, by the beginning of follicle formation (E17 in rats), active Tg synthesis is distributed homogenously among these primitive follicles and is followed by T3 and T4 formation (33,34). This more homogenous distribution of Tg is shown in Figure 2D and at higher magnification in Figure 2E. Staining for T4 shows that the hormone is localized in the colloid, covalently coupled to Tg (Fig. 2F). These events correspond closely to the appearance of functional thyrotrophs in the anterior pituitary and their secretion of TSH (35,36).
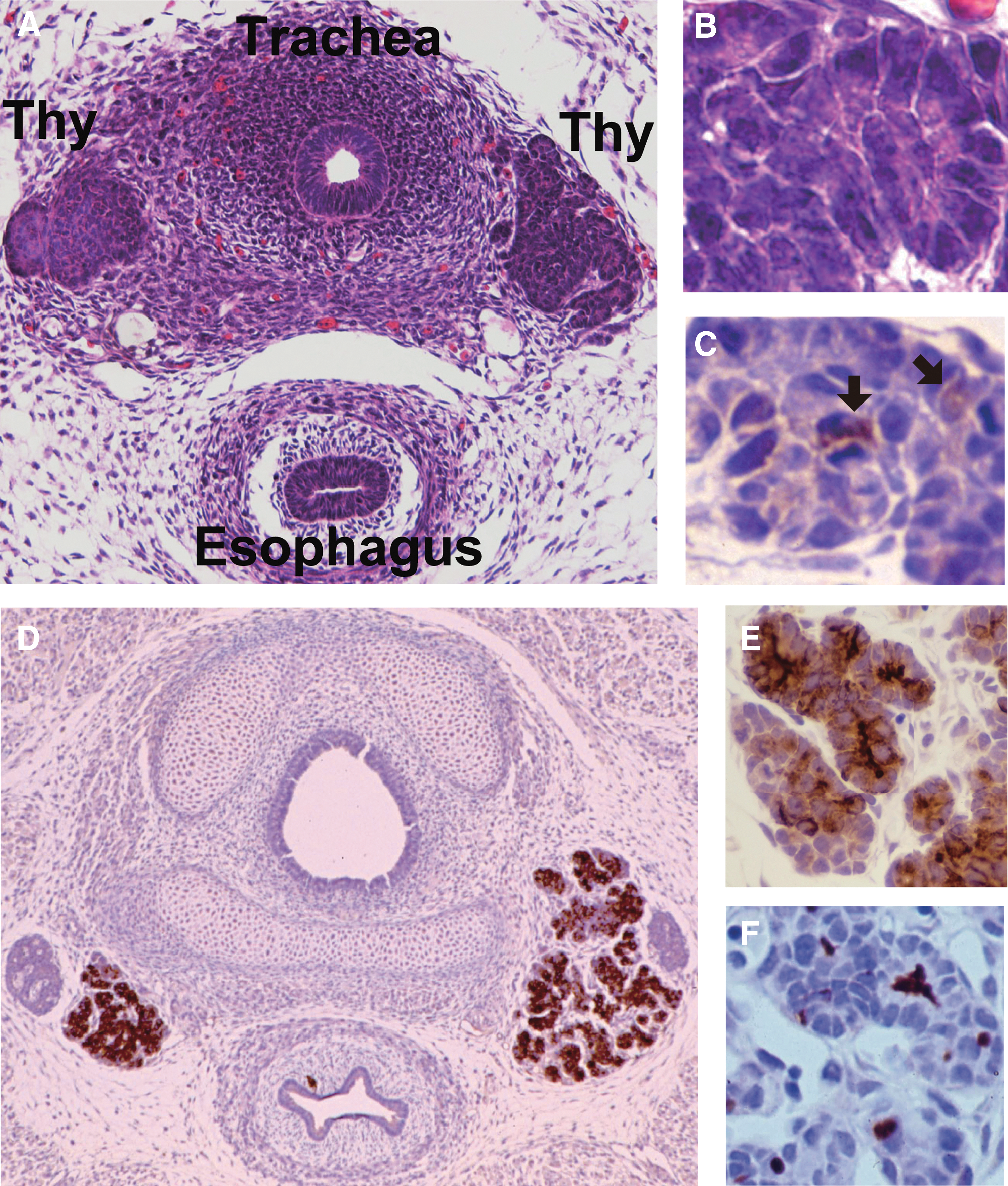
Thyroid development in the rat, illustrating early occurrence of heterogeneity.
The nature of follicular heterogeneity
The reasons for the apparent default of the thyroid to a condition of follicular heterogeneity have thus far been poorly understood. For the reasons noted above, TSH and other factors supplied from the general circulation seem to lack the ability to target individual follicles. Following the temporary period of follicular “homogeneity” and active TH synthesis, follicular heterogeneity emerges with further development as follicles begin to acquire either “active” or “inactive” phenotypes through a process that may have a genetic or epigenetic mechanism (i.e., inherited from cells in the primitive thyroid), but more likely represents the asynchronous distribution of follicles through different phases of a “follicular cycle,” as described below.
The persistence of follicular heterogeneity in the mature thyroid therefore appears to indicate the existence of an endogenous mechanism, acting independently of classical negative endocrine feedback, that is essential for maintaining the activity of individual follicles under conditions of an essentially constant TSH supply. Experimental manipulation of thyroid function with goitrogens has lent further support to this postulated intrinsic regulatory mechanism (37). Using the antithyroid drug propylthiouracil (PTU), Yi et al. showed significant decreases in follicle size and increases in epithelial height, mimicking the appearance of naturally occurring “active” follicles during application of the drug, and a gradual return to the appearance of less active follicles after the drug was withdrawn. Remarkably, despite enormous changes in TSH during the cycle of PTU treatment and withdrawal, the density of Tg present in intracellular organelles employed in Tg-trafficking was largely unchanged, leading the authors to propose that an “unknown constitutive mechanism” distinct from TSH is responsible for regulating Tg synthesis, storage, and secretion (37).
Experimental evidence for the intrinsic regulation of thyroid function by Tg
Early evidence for Tg as an intrinsic regulatory factor
Several lines of evidence have suggested that the intrinsic regulatory factor underlying the functional and morphologic heterogeneity of thyroid follicles might be the Tg molecule itself. Smeds and Anderberg (38) noted that in larger follicles, aggregates of Tg preferentially localized to the periphery of the colloid in close association with the apical thyroid cell membrane. This peripheral localization of Tg was consistent with the demonstration of apical membrane Tg-binding protein(s) on thyroid cells. The high affinity and saturable binding of these sites were suggestive of functions of Tg other than that of being a precursor for TH production, and whose uptake has generally been considered to be very low affinity or nonspecific (38 –42). Moreover, other groups observed the preferential cellular uptake of poorly iodinated, recently synthesized Tg, rather than fully iodinated and glycosylated Tg already stored in the colloid, in a seemingly counterintuitive “last come, first served” process (43 –45). These apparent discrepancies in the kinetics of resorption and refill of colloidal Tg (28,46) in concert with the heterogeneity of function and appearance of follicles in normal thyroid, led to the first detailed examination of a potential role for Tg in thyroid autoregulation, specifically in the regulation of gene expression (27,47).
Regulation of TH biosynthesis by Tg
Those initial studies, performed in a cultured FRTL-5 rat thyroid cells, demonstrated that Tg at physiologic concentrations (i.e., as measured in colloid) suppressed mRNA expression levels of several genes essential for TH synthesis, including Tg, Tpo, and Slc5a5 (NIS), whereas mRNA levels of the housekeeping gene Gapdh were unchanged (Fig. 3A–D). The possibility that the changes in gene expression are due to nonspecific effects of either altered protein concentration or osmotic pressure of the culture medium by Tg addition was examined using bovine serum albumin (BSA) and mannitol (MT) to reproduce respectively the concentration and osmolar effects of Tg (Fig. 3A–D). These agents had no effect on transcription, thus demonstrating the specificity of the observed effects to the structure of the Tg protein itself (47). Essential to the argument that the observed changes in thyroid-specific gene expression are physiologically relevant is the fact that the concentration of Tg, which comprises >95% of colloidal protein, is very high in the follicle and nowhere else in the body. In fact, Tg attains average levels as high as 110 mg/mL (48) and 250 mg/mL (49) in the rat thyroid, in a range between <1 mg/mL and >600 mg/mL for individual follicles (49). The concentrations of Tg employed in the early studies with FRTL-5 cells (0.1–10 mg/mL) (27,47,50) were chosen to reflect these previously reported follicular concentrations of Tg, not serum concentrations. Although seemingly high when compared with serum Tg, the experimental concentrations of Tg used by Suzuki et al. were in fact physiologically relevant in the thyroid and not in other healthy organs or in serum (51).
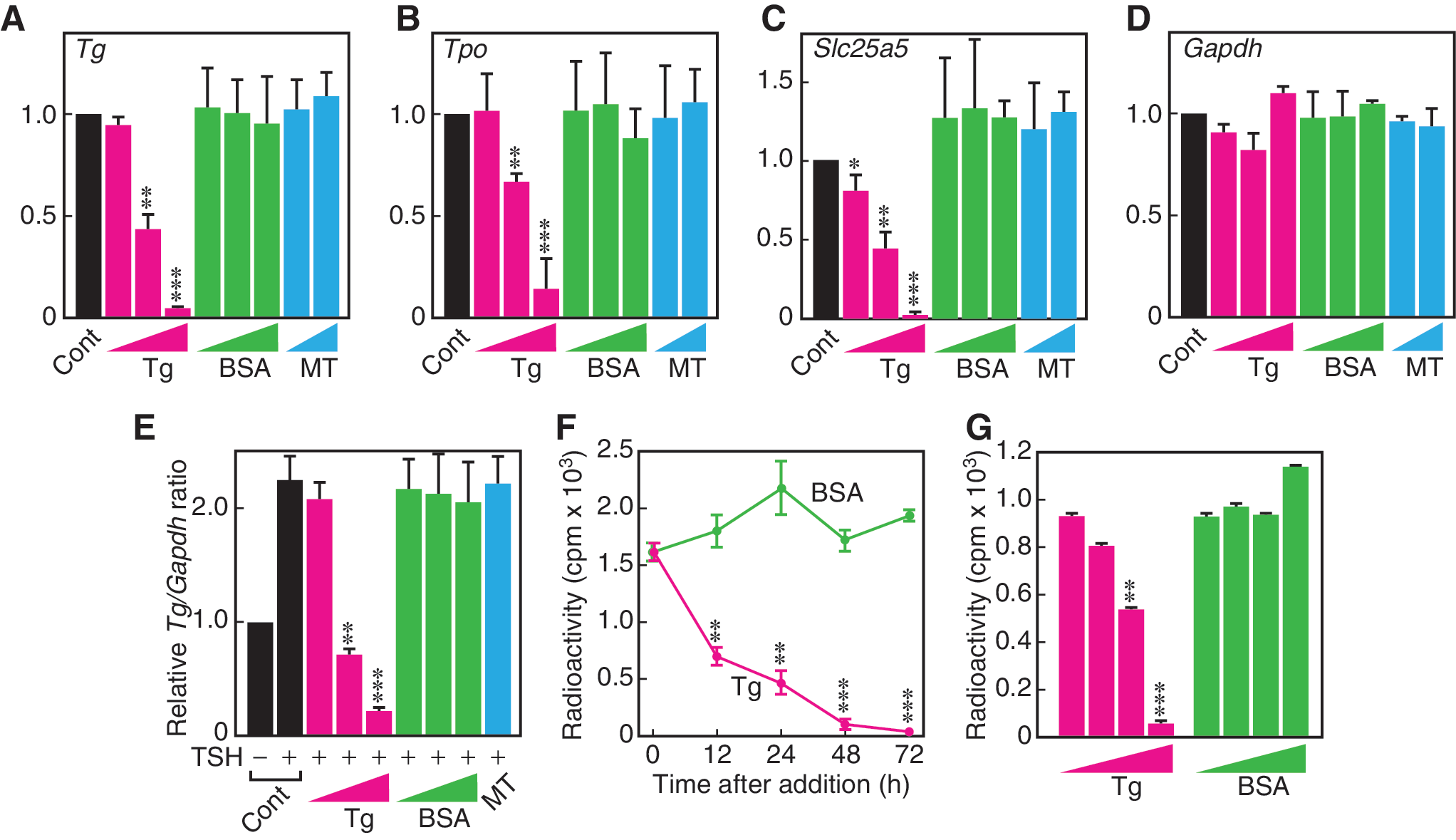
Unique effect of Tg on expression of thyroid-specific genes and iodide flux in FRTL-5 rat thyroid cells.
The critical significance of the observations described in those early studies was that Tg suppressed only those genes that are upregulated by TSH, in an apparently coordinated action to oppose the endocrine stimulation of the TH biosynthetic pathway. Specifically, whereas TSH had been demonstrated to increase Tg gene expression levels in FRTL-5 cells (9), Tg at physiologic concentrations counteracted the action of TSH, even to the point of reducing TSH-stimulated Tg mRNA expression to lower levels than in control cells receiving no hormonal stimulation (Fig. 3E) (27). If operant in vivo, a suppression of Tg gene expression by its own product, that is, the Tg protein stored in the follicle, would constitute a perfect example of short-loop autocrine negative-feedback regulation, with the important exception that the Tg signal would be received at the apical rather than basal surface of the cell (see below).
Succeeding studies of the Tg regulation of Slc5a5 (NIS) and iodide transport both in vitro and in rats lent critical support to the concept that the transcriptional effects of Tg function in vivo (50). Slc5a5, the gene encoding the basal transporter responsible for the uptake of iodide and whose expression is significantly induced in the presence of TSH (52,53), was shown to be suppressed by Tg at the promoter, mRNA, and protein levels in either the presence or absence of TSH (50). Most significantly, the ability of Tg to suppress NIS protein in FRTL-5 cells was closely associated with its ability to suppress the TSH-induced uptake of radiolabeled iodine by these cells (Fig. 3F and G) (50). Furthermore, immunohistochemical and autoradiographic analysis of sections of rat thyroid showed a clear negative correlation between the amount of Tg accumulated at the apical cell membrane of a follicular cell and the amount of radiolabeled iodine in that cell (Fig. 4) (50). Those effects were echoed in a study of PTU-treated rats, showing that a rim of Tg bound to the apical membrane of the follicular cells was associated with the suppression of Nkx2-1 mRNA expression and Tg biosynthesis in the same cells (27). Thus, Tg exhibits the same functions in vivo as already noted in vitro, namely that it acts to suppress critical elements of the TH synthetic pathway.
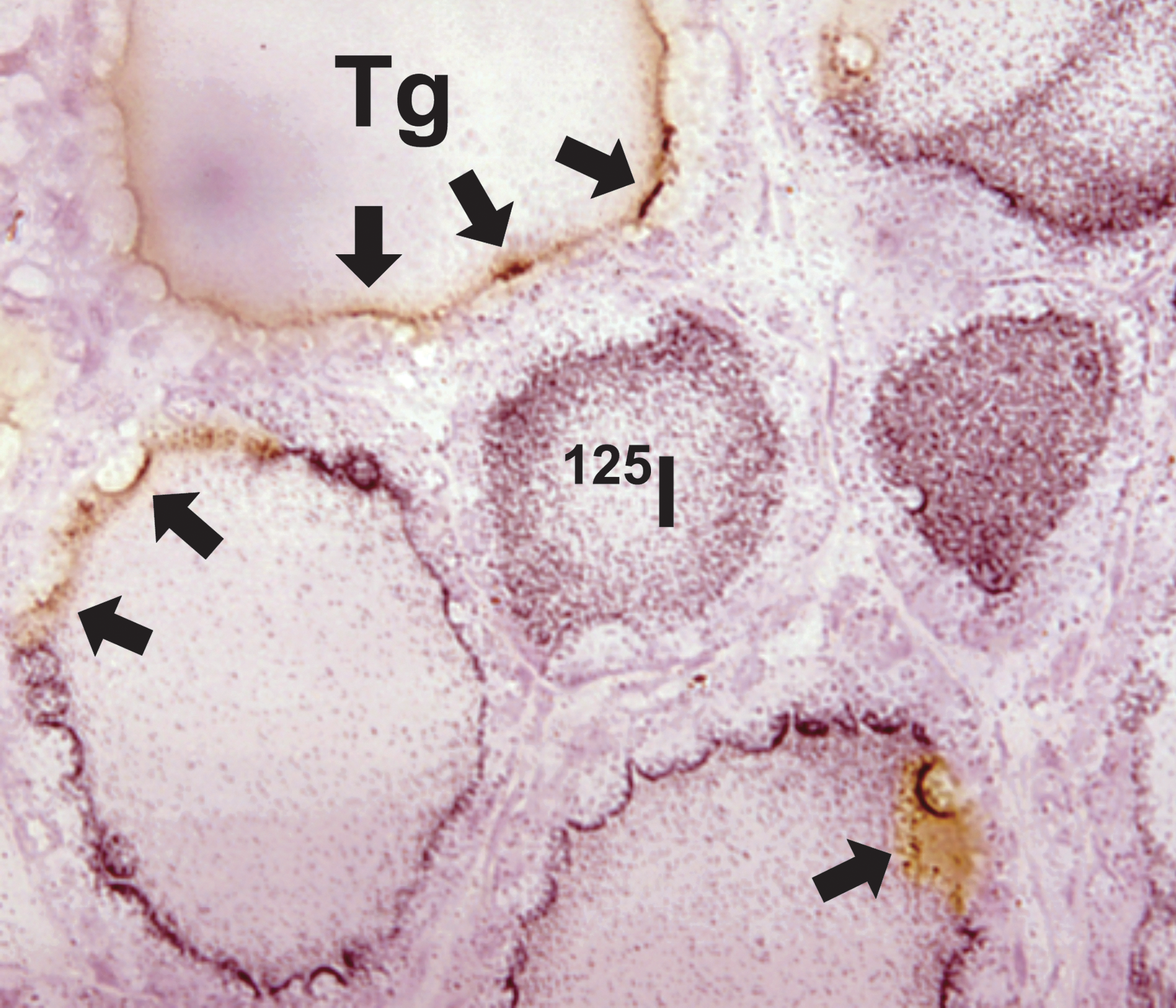
Opposing concentrations of Tg and iodide at the periphery of rat thyroid follicles. Tg distribution is identified using immunohistochemistry, and iodide distribution using 125I− and autoradiography. Arrows point to rim of Tg staining (retained after autoradiography) concentrated at the apical surface of follicular cells in areas where a paucity of silver grains suggests that iodide uptake has been suppressed.
Subsequent studies showed that Tg action is not just limited to the suppression of genes involved in TH biosynthesis, but selectively up- or downregulates those genes in an apparently coordinated action to regulate the production of TH closely (54,55). Specifically, the expression of the Pendred Syndrome Gene (PDS; Slc26a4), coding for pendrin, thought to be one of the apical iodide transport systems in thyroid follicular cells, is very low in rat thyroid tissues and in rat-derived FRTL-5 cells under basal conditions (55,56). However, it is dramatically increased in cells exposed to low levels of Tg (55,57). Remarkably, of the several thyroactive agents (T4, T3, TSH, and iodide) tested, only Tg had a significant positive effect on Slc26a4 transcript levels (55,57). Recent investigations have called into question both the uniqueness and importance of pendrin as a mechanism for iodide transport into the follicular lumen (58), although the participation of pendrin in iodide efflux overall remains very likely (59).
The most recent studies (60) have examined the Tg regulation of additional enzymes and cofactors crucial to the biosynthesis of TH, specifically the thyroid-restricted dual oxidases (DUOX1 and DUOX2) and their corresponding maturation factors (DUOXA1 and DUOXA2) whose activity generates the H2O2 required for iodide organification by TPO. In keeping with the earlier observations regarding the Tg regulation of thyroid-restricted genes, the expression of the Duox2 gene and its corresponding maturation factor gene (Duoxa2) were suppressed by Tg in a dose- and time-dependent manner, which was also observed in the suppression of other genes essential for TH biosynthesis. In contrast to Duox2 and its maturation factor, Duox1 and Duoxa1 expression were largely unaffected by treatment with Tg. This evidence is in accordance with the observation that despite sharing a >80% sequence homology (61,62), DUOX2, rather than DUOX1, is the dominant producer of H2O2 in the thyroid cell (63). Such studies not only support the concept of autoregulation of thyroid synthesis by Tg, but hint at an exquisite specificity of the process, since only one of the two closely related DUOX proteins in the thyroid is regulated by Tg. In fact, the suppression of DUOX2 alone was sufficient to reduce the H2O2 production in FRTL-5 thyroid cells significantly and dramatically (60). This suggests that despite the apparent redundancy of function of the two DUOX isoforms, Tg in vivo would nevertheless reduce H2O2 production sufficiently (together with the reduction in TPO) to suppress the iodide organification required for TH synthesis.
Regulation of transcription factors
In regard to how the genes involved in TH production are coordinated by the action of follicular Tg to regulate circulating TH levels, both the initial report of Suzuki et al. and subsequent studies (47,50,55,57) demonstrated that the ultimate action of Tg occurs at the level of gene transcription, rather than on mRNA stability, protein translation, or other actions downstream of promoter activation. Further investigation showed that the mRNA expression of three “thyroid-restricted” transcription factors—Nkx2-1, Foxe1, and Pax8—responsible for increasing Tg after TSH stimulation were also dramatically reduced, whereas the expression levels of several ubiquitous transcription factors that are also required for the transcription of thyroid-specific genes—Sox4, Tsep1, and Ssbp1—were unaltered (27,47).
Pax-8, a paired box transcription factor that binds to an element overlapping the Nkx2-1 binding site in the promoter of thyroid-specific genes (64), is usually considered a thyroid-restricted transcription factor, essential both in thyroid morphogenesis and endocrine function (65,66). However, unlike TTF-1 and TTF-2, Pax-8 is actually expressed at relatively high levels in several nonthyroid tissues/cell types, including the glomerular mesangial cell, in which it was observed that at sufficiently high concentration (1 and 10 mg/mL), Tg significantly suppresses Pax8 transcript levels (67,68)
The most comprehensive study to date of gene regulation by Tg used DNA microarray analysis in FRTL-5 cells to examine Tg influence on the expression of genes in a number of intracellular functions including hormonal signaling pathways and cell division (69). In addition to the thyroid-specific and other genes already discussed, Tg was found to have a profound effect (primarily stimulatory) on the mRNA expression of such genes as prostaglandin E synthase (Ptges; more than ninefold) and fibronectin (Fn1; more than sevenfold) and on genes involved in cell cycle regulation, including cell division cycle associated 7 (Cdca7), early growth response 1 (Egf1), and Runx1 in the absence of TSH, insulin, and serum. These findings, although quite preliminary, provide a hint as to the more global functions of Tg signaling in the thyroid cell, including regulation of subcellular matrix expression, and of cell cycle-related genes, the latter of which could help to explain the observed effects of Tg on cell proliferation that are described below.
Regulation of cell proliferation
Following its recognition as a regulatory factor in TH synthesis, additional actions of Tg in the thyroid gland have begun to emerge, most notably its marked stimulation of follicular cell proliferation (69,70). Ironically, some of the first demonstrations of a direct effect of Tg on cell proliferation were performed with mink lung cells, a cell type that has no obvious physiologic relationship to the TH precursor, but in which Tg was shown to block the TGF-β-induced inhibition of DNA synthesis (71). A subsequent study of Tg effects on proliferation in the renal mesangial cell, a cell that like the follicular cell expresses high levels of Pax-8, also showed a significant increase in cell growth in response to Tg (68).
FRTL-5 cells in culture responded to Tg much as mesangial cells did, with a significant elevation in both growth rate and 3H-TdR uptake, in contrast to several cultures of cancer-derived cells that exhibited no change in proliferation with Tg (70). Most significantly, the stimulatory effect of Tg on thyroid cell growth was pronounced, even in the absence of insulin or insulin-like growth factor-1 (IGF-1). In contrast, TSH stimulation of cell growth required insulin or IGF-1 as cofactors in order to achieve an increase in cell proliferation comparable to what was achieved with Tg alone (70). These results suggest the interesting possibility that Tg can act as an independent promoter of thyroid cell proliferation. Further investigation using three-dimensional follicular structures will be needed to determine the functional significance of the in vitro observations. In what appears to be a complex process of hormonal counter-regulation in the thyroid cell, the combination of Tg with TSH (or downstream activators of TSH signaling such as forskolin and dibutyryl cAMP) actually decreased FRTL-5 growth parameters compared with Tg alone (70). Although apparently counterintuitive, inhibition of Tg-stimulated cell proliferation by TSH could be critically important in regulating growth in follicles that cycle through alternating phases of “Tg” and “TSH” dominance (see below). Later studies by this group (69) showed that the response to Tg in thyroid cell proliferation was biphasic in a manner reminiscent of the response of pendrin expression to Tg, with a several-fold increase in cell number occurring with moderate levels of Tg (5 mg/mL), and a return to basal cell number with high Tg concentrations (20 mg/mL). Together, the TSH sensitivity and narrow concentration range of the Tg-induced increase in thyroid cell proliferation suggest that thyroid cell number is effectively coupled to changes occurring in colloid volume.
Other actions
The early investigations into Tg as a regulator of thyroid-specific gene expression also examined effects of Tg on the expression of major histocompatibility complex (MHC) class I, a gene of ubiquitous distribution but of particular interest in the thyroid, since this gland is a frequent target of autoimmunity (72,73). The findings (47) showed that MHC class I mRNA levels were significantly increased (about twofold) by Tg at a concentration of 1 mg/mL in rat thyroid FRTL-5 cells, but were unchanged in several nonthyroid cell lines, suggesting that a Tg effect on self-recognition and autoimmunity is limited to the thyroid. As with Slc26a4 expression, the effect of Tg on MHC class I transcription, contrary to all the genes suppressed by Tg, is essentially a biphasic one, with maximum response occurring at moderate Tg concentrations (1 mg/mL) rather than the highest ones (10 mg/mL). This might suggest that the mechanisms/pathways leading to gene activation by Tg are distinct from those leading to gene suppression.
Another gene of more ubiquitous expression than most of the Tg-regulated genes in the thyroid cell is Vegf, whose expression levels were found to be reduced in cultured FRTL-5 cells by Tg in parallel with Slc5a5 and Tg mRNA in a time-dependent fashion (50). Like Tg, Tpo, and Slc5a5 but unlike Slc26a4, Vegf gene expression is regulated by TSH in thyroid cells (74). Hence, its regulation by Tg may share common pathways with the genes in the TH pathway that are under negative control by Tg. Downregulation of thyroid cell VEGF by Tg in vivo would be expected to alter the function of the microvasculature surrounding the follicle, but whether the principal effect is endothelial cell growth or permeability or some other specific action in the follicular blood supply remains in question (75). Gérard et al. (76) have described “angiofollicular units” in the thyroid gland in which follicular morphology is closely linked to the appearance and functioning of the microvasculature surrounding it, and suggest that vasodilatory factors such as VEGF produced in thyroid follicular cells may be involved in this coordination of epithelial and endothelial morphology and function. The suppression of VEGF expression by Tg and the variation between follicles in the concentration and iodination state of Tg offers one possible explanation for both the occurrence of angiofollicular units and their heterogeneity of appearance and function throughout the thyroid.
Potential mechanisms for Tg autoregulation
Cell membrane receptors for Tg
The least well understood aspect of thyroid autoregulation—and one of the main factors weighing against the widespread acceptance of the concept—is the precise mechanism by which Tg transduces its action on the thyroid follicular cell. Unlike TSH, which binds with high affinity to a singular member of the family of G-coupled transmembrane proteins (77), Tg binds to several unrelated proteins on the surface of thyroid cells, none of them identified strongly with intracellular signal transduction. Moreover, these Tg-binding proteins, in comparison with the TSHR, are not particularly selective as to the ligand to which they bind. Most of them bind to other large glycoproteins fulfilling the binding criteria (e.g., the possession of a terminal galactose residue) with a comparable affinity to Tg (78,79).
Another major difference between the proposed regulation of the thyroid by Tg and regulation by circulating TSH is that whereas TSH signal transduction takes place at the basolateral surface of the follicular cell, Tg control of thyroid function is an “apical” phenomenon, with receptors/ transduction systems in direct contact with high concentrations of Tg in the follicular lumen. The apical localization of the putative Tg signal transduction mechanism also distinguishes follicular autoregulation by Tg from more conventional paracrine and autocrine regulation, whose signals are confined to targets in the basolateral rather than apical membrane of the cell.
The Tg-binding protein receiving the most support for transducing the effect of Tg on gene expression is an asialoglycoprotein receptor (ASGPR), a member of a class of receptors that delivers receptor-bound ligand to intracellular compartments (80). The thyroid ASGPR is related to the ASGPR of the liver, which acts in a nonselective manner to clear circulating glycoproteins from the plasma (80,81). In the thyroid, however, the ASGPR is on the apical rather than basal aspect of follicular cells, where it functions to bind newly synthesized Tg and transport it via a process of receptor-mediated endocytosis to various intracellular compartments including endosomes and ultimately to lysosomes for degradation (81,82).
Indirect evidence supporting a role for thyroid ASGPR in the regulation of thyroid function by Tg includes the abrogation of Tg suppression of Nkx2-1 promoter activity by preincubation with an antibody against the ASGPR, and a close correspondence between the ability of various multimeric forms of Tg (27S, 19S, and 12S) both to bind the ASGPR and to influence the activity of thyroid-specific genes (83). Moreover, poorly sialylated and poorly iodinated Tg localized at the periphery of the follicular colloid has both a greater affinity for the ASGPR and a greater effectiveness in altering the expression of thyroid-specific genes than does normally sialylated Tg (54). Despite the apparent linkage of the ASGPR to the transcriptional effects of Tg, however, there are as yet no clear data linking Tg binding by the apical ASGPR to a process of intracellular signal transduction (39,42,83,84). Weighing against a link between Tg autoregulation and the ASGPR, moreover, is the finding that no abnormality in either thyroid function or morphology has been noted in ASGPR-deficient mice (Suzuki K and Kohn LD, unpublished observation).
In addition to the ASGPR, a number of other Tg receptors have been proposed or identified. These include megalin (gp330), a protein that mediates the transcytosis of Tg through the follicular cell (85), an N-acetylglucosamine receptor (86), a protein disulfide isomerase, and several low-affinity receptors (79,87). However, it is unlikely that a high affinity receptor is needed to bind Tg, the most abundant protein in the colloid. Much of the uptake of Tg from the colloid occurs through nonspecific fluid-phase micropinocytosis, which unlike Tg receptor binding is essentially nonsaturable. Given the very high concentrations of Tg in the colloid, it has been argued that nonspecific uptake of Tg rather than receptor-mediated endocytosis is actually the prevailing mode of Tg entry into the thyroid cell in vivo (85). Several studies (88,89) have demonstrated the existence of invaginated cholesterol and spingolipid-rich microdomains known as caveolae in the apical domain of thyroid cells that could be involved in the nonspecific micropinocytosis of Tg. These microdomains—actually a form of lipid raft—also contain one or more members of a family of proteins known as caveolins (caveolin 1-, 2-, and −3) that are involved in the budding process in a manner similar to the budding of clathrin-coated vesicles, whose involvement in Tg endocytosis has also been noted (90).
Most intriguingly, a recent study has demonstrated (89) that alterations in thyroid morphology and function in mice lacking caveolin-1 (Cav-1−/−)—an apical membrane protein essential to the formation of caveolae (91,92)—showed a striking similarity to what would be predicted with a reduction or abrogation of a Tg regulatory signal in the follicular cell. These changes included distended cisternae of the rough endoplasmic reticulum (rER) resulting in increased cellular height, increased intracellular accumulation of Tg, DUOX, and TPO, and reduction or absence of Tg in the follicular lumina (89).
An alternative explanation of the close correspondence of Tg and its binding proteins with resultant changes in thyroid gene transcription is that it is unrelated to receptor-mediated signal transduction, but is instead due to the Tg or Tg fragments transported into the cell. Greater concentrations of colloidal Tg would result in greater numbers of intracellular Tg fragments available to perform specific autoregulatory functions. Such observations call for further investigation to determine the nature of the proteins and intracellular transduction pathways that could account for those similarities.
Signaling used for Tg-induced thyroid cell growth
Although a definitive cell surface receptor linking follicular Tg to transcriptional regulation remains elusive, elements of several signal pathways linking apical Tg to changes in follicular cell function have been described. Remarkably, the activation of the adenylate cyclase/cyclic adenosine 3,5-monophosphate (cAMP)/protein kinase A (PKA) signal pathway, so critical to the regulation of thyroid cell function by TSH, is not one of them. Noguchi et al. (70) showed that at levels resulting in significant increases in the proliferation of FRTL-5 thyroid cells, Tg had no effect on cAMP levels. Moreover, treatment of the cells with the PKA inhibitor H-89 did not alter the positive effect on FRTL-5 growth parameters. On the contrary, activation of the TSH/cAMP pathway actually inhibited the induction of cell growth by Tg in an action opposite to its widely recognized function as the principal effector of follicular cell proliferation (70).
Unlike cAMP/PKA inhibition, suppression of the phosphatidylinositol 3-kinase (PI3K)/Akt signaling pathway was found to have a profound inhibitory effect on the action of Tg to stimulate FRTL-5 cell proliferation (69,70). Acting downstream from insulin/IGF-1 in the thyroid follicular cell, Akt activation is known to be a key intermediate in the stimulation of cell growth by the combined action of TSH and insulin (70,93). Therefore, it is possible that both Tg and insulin/TSH stimulation of thyroid cells employ a common pathway to increase cell proliferation downstream of PI3K/Akt activation. However, upstream of Akt, Tg signaling cascades remain undefined, and are likely distinct from those of other effectors leading to increased thyroid growth. Taken together, the evidence thus far suggests the existence of hitherto unknown interaction and/or crosstalk of signaling cascades between Tg and TSH that will make the study of Tg signaling under physiologic conditions (i.e., in vitro under TSH stimulation, or in vivo) a significant challenge.
An additional intracellular signal pathway employed by Tg—and unlike PI3K, one not shared with TSH—is the activation of c-Raf/MEK/ERK leading to increased proliferation of the follicular cell (69). Strong support for the relevance of this Tg-signal pathway was obtained from studies showing the phosphorylation of c-Raf, MEK1/2, and ERK1/2 after treatment with Tg, and a significant suppression of Tg-stimulated BrdU incorporation into DNA in the presence of the MEK1/2 inhibitor, PD980159 in FRTL-5 cells. In marked contrast, TSH stimulation of BrdU incorporation was unaffected by pretreatment with PD98059 (69).
The thyroid follicular cycle: a model for negative-feedback Tg autoregulation of thyroid function
Despite the many unanswered questions remaining, it is useful to fit the accumulated data on the transcriptional activity of Tg into a plausible model of thyroid autoregulation. That model must incorporate the following observations and concepts. First, Tg is secreted by the thyroid cell into the follicular lumen via a process of regulated (nonconstitutive), merocrine secretion similar to that of many other secretory epithelial cell types (94,95). Second, upon stimulation by TSH, Tg is reabsorbed by endocytosis/pinocytosis or phagocytosis (rodents only) to form endocytic/pinocytotic vesicles, or phagosomes, respectively (96 –98). Third, the volume and concentration of Tg in each follicle therefore is a result of the balance between the secretion from and reabsorption of Tg by the surrounding follicular cells. Detailed analyses of the kinetics of Tg secretion and absorption, however, indicate that follicular Tg concentration after TSH stimulation is not as readily equilibrated as had been previously assumed (99,100), but instead requires a considerable amount of time to compensate fully for the removal of colloidal Tg by replacing it with newly synthesized Tg (Fig. 5) (101).
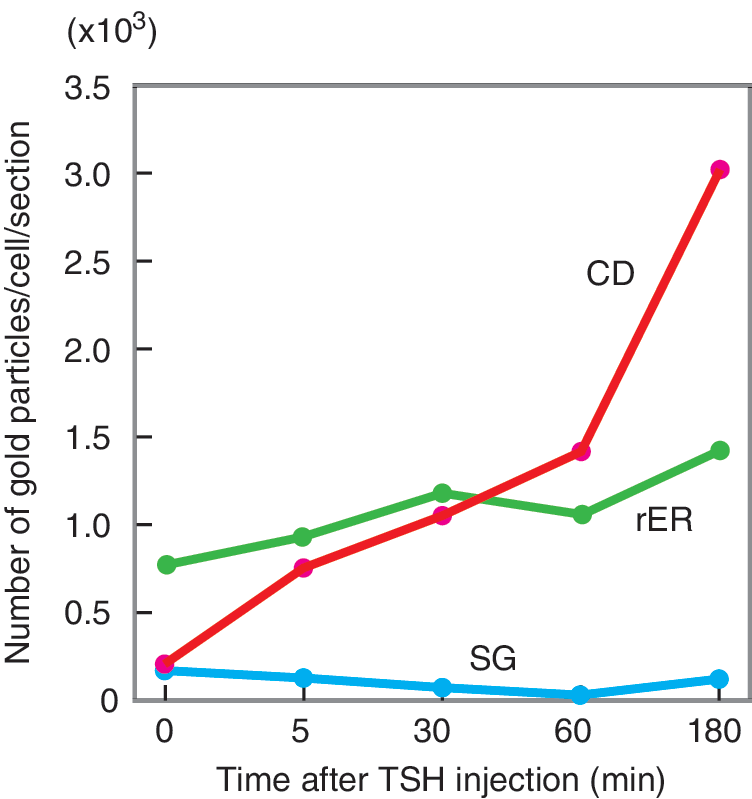
Kinetics of subcellular immunogold-labeled Tg distribution after TSH. The number of gold-labeled particles was counted in each of several subcellular organelles. CD, internalized colloid droplets; rER, rough endoplasmic reticulum; SG, secretory vesicles (granules).
A possible reason for the discrepancy between the kinetics of Tg production and internalization is that colloid resorption and subsequent Tg degradation are rapid processes necessitating relatively simple physical and chemical reactions in order to be carried out efficiently to maintain normal serum TH levels, whereas the refill of the follicular lumen with secreted Tg requires a much longer time due to the lengthy processes of transcription, translation, and posttranslational modification of Tg that must occur first. As a result, Tg concentration in the single follicle does not remain stable, but instead changes concentration over a period of time. Moreover, as discussed above, neighboring follicles are not synchronized in their functional states, and thus under normal circumstances differ greatly both in size and in relative activity (19,20).
Given that Tg contents among follicles are highly variable (Fig. 1B) (48,49,101 –103) at any given time regardless of the endocrine status of the individual, high Tg follicles will experience enhanced Tg resorption while low Tg follicles nearby will have a dampening of Tg resorption as they refill their depleted Tg stores. This phenomenon has actually been demonstrated in vivo in a report showing that newly synthesized, hormone-poor Tg recently delivered to the follicle lumen (i.e., in Tg filling phase) is prevented from immediate reuptake (97). From a teleological point of view, it would make sense for a given follicle to reduce or suspend the resorption of colloid during the period of time in which it is synthesizing Tg and refilling the follicular lumen, since endocytosis or pinocytosis of colloid low in Tg content would constitute a waste of cellular energy in the vital process of maintaining TH at homeostatic levels. For maximum efficiency of TH production, Tg resorption should take place in those follicles storing a large amount of Tg, and it should be suspended during refilling of the colloid with Tg, even in the presence of high circulating TSH levels. The underlying mechanisms for this observation were unclear at the time of that report, but evidence of regulatory activities of the Tg molecule made in the intervening years suggests that Tg is critical for signaling the changes in Tg synthesis and resorption that create a “cycle” of the thyroid follicle, with follicles throughout the gland in different phases of that cycle at any given time accounting for follicular heterogeneity.
Figure 6A illustrates a model for the potential functional cycle of a thyroid follicle. At the upper far left is a follicle in which colloidal Tg concentration is low (light color), transcription of substrate (Tg), enzymes (TPO, DUOX2, and DUOXA2), and transporters (NIS and Pendrin) involved in TH synthesis are high (reflecting maximum expression of essential transcription factors: Nkx2-1, Foxe1, and Pax8), and active uptake of iodide takes place (red arrows) as these processes are released from suppression by apical Tg. This corresponds to the synthesis phase in the graph below, and is indicated as follicle I in the photomicrograph of thyroid immunostained for Tg (Fig. 6B). Follicle I exhibits a low concentration of Tg in the colloid, and conversely, a clearly visible intracellular Tg staining (Fig. 6B, arrows) reflecting new Tg synthesis in the rER and Golgi apparatus (28,37,101). In such a follicle, still low in colloidal Tg (and, more importantly, low in iodinated Tg), endocytosis or pinocytosis of the follicular contents would be unable to import sufficient iodinated Tg into the cell to achieve an efficient production of TH. Therefore, a suppression of Tg breakdown and TH release likely dominates at this point. Cell proliferation is also low in the synthesis stage (70). Extrapolating from in vitro results, we speculate that as a higher (but still below maximal) concentration of Tg accumulates in a follicle (Fig. 6B, follicle II), cell proliferation reaches its maximum (70), possibly in order to form new follicular epithelium as Tg synthesis and secretion lead to a larger colloid volume.

To the right of this follicle in Fig. 6A is one in which colloidal Tg and iodinated Tg reach maximal levels (dark color) as a result of processes occurring in the preceding phase. This follicle corresponds to the storage phase in the graph below. This stage of maximal Tg filling of the follicular lumen is also shown in the photomicrograph of a follicle (Fig. 6B, follicle III) with heavy colloidal Tg staining but reduced staining in the follicular epithelium. After a sufficient amount of Tg has accumulated in the follicle, all genes and cellular activities involved in TH production become suppressed by the negative feedback regulation of Tg (white arrows) (27,47,50,55,57). We suggest that endocytosis and hormone secretion can now take place most efficiently. This process is represented as outwardly directed red arrows in the follicle (Fig. 6A, top right), representing TH secretion, and is continued throughout the secretion phase in the graph below. Then, as hormone-laden Tg is endocytosed and degraded in lysosomes to release TH, colloidal Tg concentrations fall to levels at which negative feedback suppression of gene transcription can no longer be sustained, and in the presence of a more-or-less constant stimulation by TSH, expression of genes involved in TH biosynthesis begins to rise, marking the beginning of a new synthetic phase (Fig. 6A).
The curve representing fluctuations of follicular TH over the course of a single cycle (Fig. 6A) depicts a longer synthetic phase than secretion phase, reflecting the dependence on transcriptional events in the former and post-translational events in the latter, as was described above. However, the lengths of the phases shown—and of the entire cycle—should not be taken too literally. The duration of each will necessarily vary due to many factors, including the size of the follicle at the beginning of a cycle, the total amount of iodide uptake in a follicle, and even the overall metabolic status of the individual. A more definitive picture of the temporal changes in follicular form and function occurring throughout the postulated cycle will emerge when it becomes possible to observe live images of follicular activities in vivo.
Conclusions
Tg, long-regarded solely as the high molecular weight precursor for TH formation, has now been demonstrated to possess intrinsic regulatory properties unrelated to the TH bound covalently within it. Furthermore, the direct effects of Tg on thyroid cell function point to the existence of an intrinsic negative feedback regulation of TH production complementing the extrinsically controlled HPT axis to exert exquisite control over circulating TH levels. In addition, the long-standing conundrum of the heterogeneity of thyroid follicle morphology and function in the face of the presumably homogeneous exposure of thyroid tissue to TSH and other hormones can now be explained as a consequence of the autoregulatory role of Tg.
Now that the phenomenon of Tg regulation of gene transcription and thyroid function has been established, the emphasis must shift to describing the precise mechanism or mechanisms underlying the observed effects of Tg. It will be especially important to clarify the interaction with and counteraction of Tg by known thyroid regulators, for example TSH, insulin/IGF, and iodide. Detailed analyses of these complex interactions will require the use of models that preserve a greater degree of the normal thyroid cytoarchitechture such as three-dimensional thyroid cell culture systems that have been employed in several previous studies (104 –106). A complete description of the regulatory action of Tg would include both an identification of the cell surface and/or intracellular recognition systems that are involved, and also which domain(s) of the Tg molecule are recognized by those systems to generate a regulatory signal.
Further investigations proceeding from the surprising finding that Tg is an autoregulatory molecule should allow a better understanding of how thyroid homeostasis is maintained, and possibly how that homeostasis is disrupted in thyroid disease (93).
Footnotes
Acknowledgments
The authors thank Ms. Rebecca Sellitti for her editorial assistance. This work was supported in part by a Grant-in-Aid for Scientific Research from Japan Society for the Promotion of Science (#33802400 to K.S.).
Author Disclosure Statement
The authors have no financial agreements or conflicts of interest to disclose.