
Abstract
Background:
The cribriform morular variant of papillary thyroid carcinoma (CMVPTC) is a rare subtype of papillary thyroid cancer that occurs most often in association with the familial adenomatous polyposis (FAP) syndrome.
Patient findings:
A 18-year-old woman presented with recurrence of PTC in her neck. She had a prior diagnosis of FAP syndrome. Review of her original pathology slides reclassified the case as a CMVPTC. The tumor was examined for the four most common mutations found in PTC: BRAF, RET/PTC, RAS, and PAX/PPARγ.
Summary:
The molecular alterations associated with CMVPTC involve the WNT signaling pathway but are incompletely understood. When CMVPTC is associated with the FAP syndrome, a germline adenomatous polyposis coli (APC) gene mutation is almost always detected. For the initiation of oncogenesis however, one or more additional molecular alterations must occur, such as a new somatic mutation in the APC gene (biallelic inactivation), somatic mutations in the β-catenin (CTNNB1) gene, or gene–gene interaction (epistasis). To date, of the mutations commonly associated with PTC, only RET/PTC mutations have been reported in CMVPTC. We report a FAP-associated CMVPTC tumor with atypically aggressive features harboring a RAS mutation and review the molecular mechanisms associated with this interesting PTC subtype. The literature was reviewed using MEDLINE (included case presentations, original research, and reviews).
Conclusion:
We report here the first RAS mutation detected in an FAP-associated CMVPTC tumor.
Introduction
F
Thyroid cancer is reported to have a 1%–2% prevalence in FAP, but this may be an underestimation (4,5). The vast majority of FAP-associated TC cases are papillary thyroid carcinoma (PTC); 20%–40% of these PTCs are the extremely rare cribriform morular variant of PTC (CMVPTC) (6,7). The reason for the strikingly high frequency of this otherwise rare subtype of PTC in FAP is not clear, but the answer may lie in the unique molecular composition of this tumor.
We report here a patient with CMVPTC to illustrate the most relevant clinical and histological aspects of this rare tumor. We provide a review of the available literature on FAP-associated CMVPTC, specifically focusing on molecular markers and mechanism. Furthermore, we report the first case of CMVPTC associated with a K-RAS mutation.
Patient
V.A. was diagnosed with FAP at age 18 years after her father was diagnosed with the same condition a few months earlier. Two other family members were subsequently diagnosed with FAP. Prophylactic colectomy was performed with no evidence of malignancy on pathologic examination. Two years prior to the diagnosis of FAP, the patient had been diagnosed with thyroid cancer. Thyroidectomy and pathologic examination revealed a right-sided, 2.4 cm, tall-cell variant of PTC with invasion of surrounding thyroid tissues, and a second “histologically similar” 1.1 cm tumor in the left thyroid lobe. Remnant ablation with 58 mCi of 131I was performed. Posttreatment radioiodine scan results are unknown to us.
At age 20 years, the patient was diagnosed with multiple desmoid tumors as well as a recurrence of PTC in the neck. While most fine-needle aspiration (FNA) smears from the recurrent right neck lesion showed features consistent with classic PTC, one sample showed a cluster of fasciculated spindled cells, unusual for conventional PTC (Fig. 1). The histology slides from her first thyroid surgery were thus requested for re-examination. These samples showed a combination of multiple growth patterns, including fascicular spindled cells and squamoid islands (morules), scattered through the tumor (Fig. 2). Immunohistochemistry showed a diagnostic nuclear staining positive for β-catenin (Fig. 3A). The morules exhibited prominent nuclear staining for BCL-2 and cytoplasmic staining for CD-10 (Fig. 3B,C). The entire tumor was negative for thyroglobulin (Fig. 3D). The case was reclassified as CMVPTC. Subsequently, the tumor was examined for the four most common mutations found in PTC, namely BRAF, RET/PTC, RAS, and PAX/PPARγ. A novel K-RAS mutation c. 181C>A (p. Q61K) was the only mutation detected.
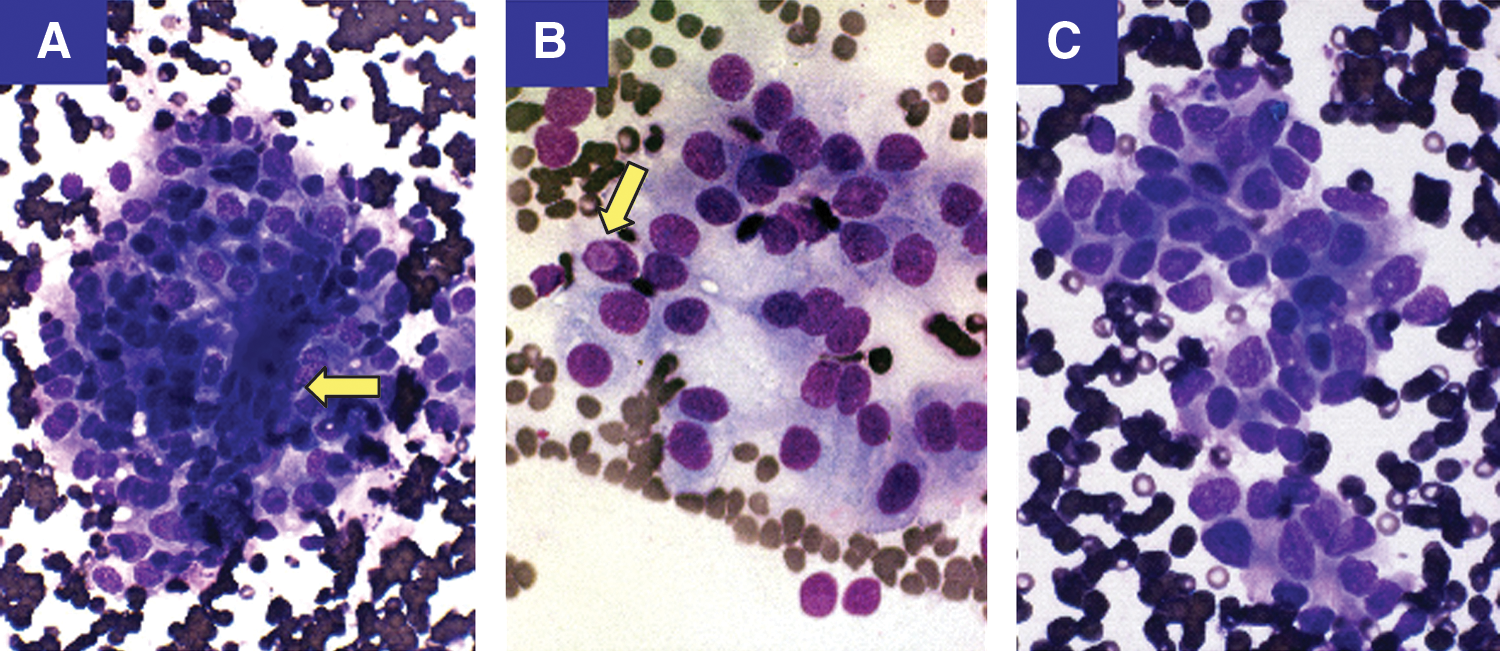
Cytologic features of cribriform morular variant of papillary thyroid carcinoma (CMVPTC) metastatic to lymph node. The aspirate shows a cluster of fascicular spindled cells (arrow)
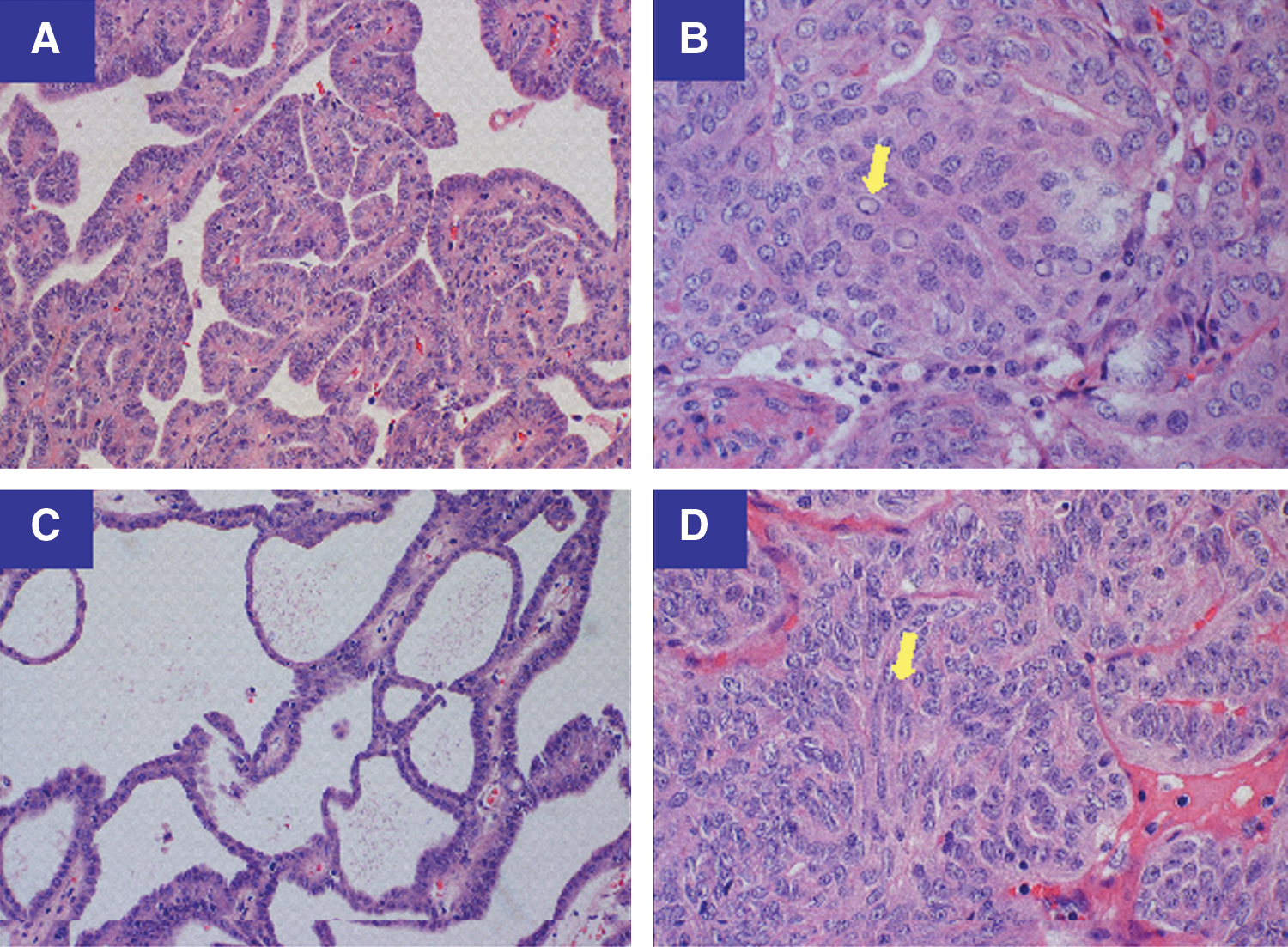
Histologic features of CMVPTC. CMVPTC exhibits two distinct histological components. The epithelial component presents a combination of papillary
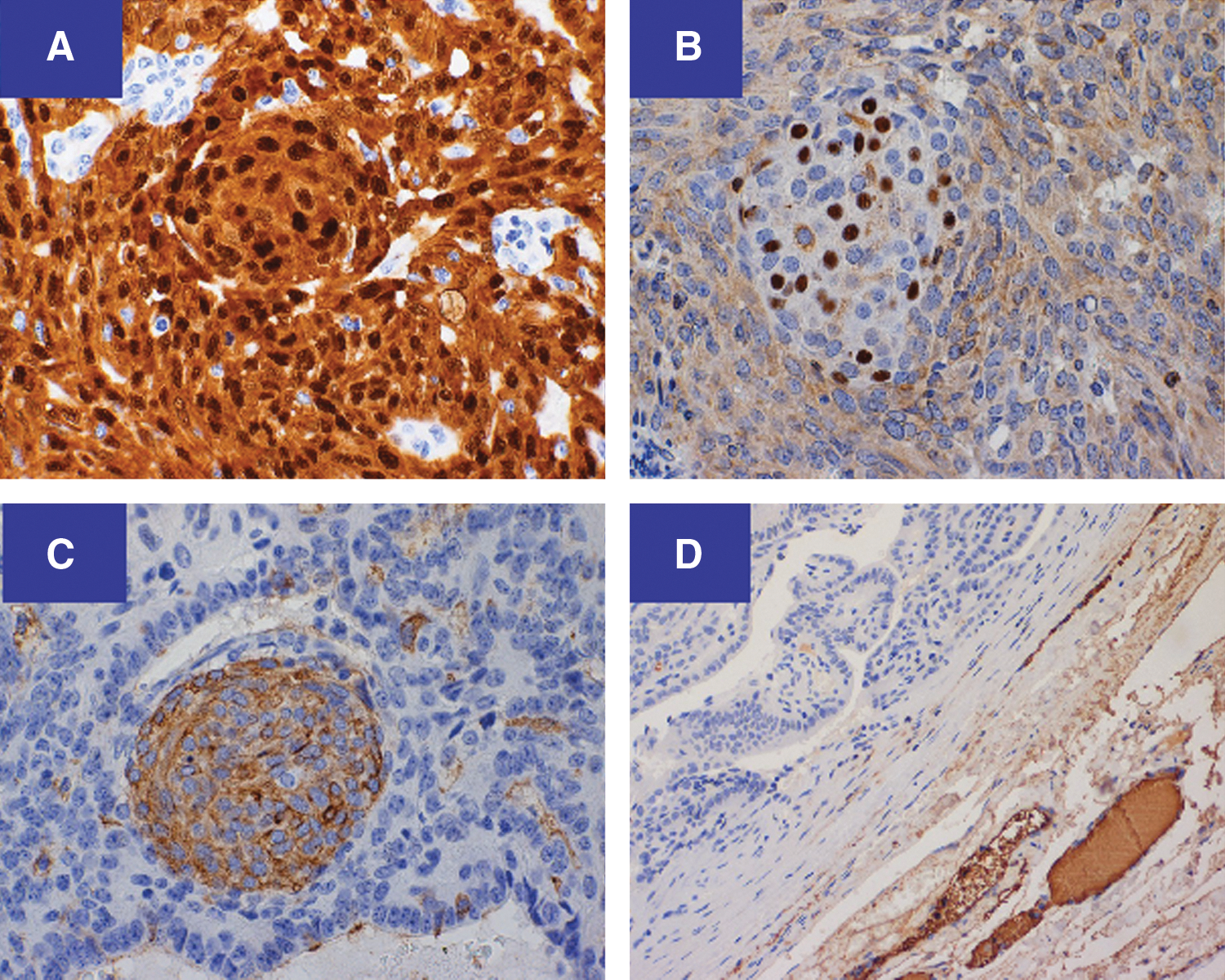
Immunohistochemical features of CMVPTC. The entire tumor shows cytoplasmic as well as diagnostic nuclear staining for β-catenin
Methods
Cytologic, histologic, and immunohistochemical examinations
FNA was performed and smears were air-dried and stained with a modified Wright-Giemsa method (Diff Quik, Siemens Healthcare Diagnostics, Inc.). The thyroid tumor specimen was fixed in zinc-buffered formalin, processed with gradient alcohols and xylene, and paraffin embedded. Four-micrometer-thick sections were stained with hematoxylin and eosin. Immunohistochemical studies were performed on paraffin sections using a panel of antibodies against TTF-1 (1:00 dilution; Cell Marque), β-catenin (Ab clone 14 at 1:80 dilution, Cell Marque), BCL-2 (Ab clone 124 at 1:100 dilution; DAKO), CD-10 (Ab clone 56C6 at 1:100 dilution; Novocastra Leica Biosystems), and thyroglobulin (Ab clone 2H1H6E1 at 1:200 dilution; Biocare). We employed the Benchmark XT detection system (Ventana Medical Systems). Samples were not pretreated. We used the heat-induced epitope retrieval technique (pressure cooker) using citrate buffer (pH 6.0) to pretreat samples for CD-10 and BCL-2 detection. CD-10 antigen was detected using the Power Vision+Poly-HRP (Novocastra) detection system and the BCL-2 oncoprotein was detected using the HRP Detection system (Cell Marque).
Molecular analysis
Molecular analysis was performed at Quest Diagnostics Nichols Institute using unstained paraffin-embedded sections. Allele specific oligonucleotide polymerase chain reaction (PCR) was used to detect BRAF mutations, specifically looking for the presence of V600E, V600K, or K601E. For detection of RET/PTC (either RET/PTC1 or RET/PTC3) and PAX/PPARγ rearrangements, total RNA was reversed-transcribed, with cDNA quantification (relative to an ABL1 control transcript) of any rearranged transcripts performed using fluorescent real-time PCR on an ABI PRISM 7900HT sequence detection system. Detection of RAS (H, K, or N) mutations at codons 12, 13, or 61 was performed by pyrosequencing on a PyroMark Q96 MD platform.
Discussion
Clinical
This case represents an example of CMVPTC presenting in the context of FAP. The typical patient is a young woman, as this malignancy has a striking female preponderance (1) of 17:1, and presents earlier than classic PTC, at a mean age of 27.7 versus 45 years (7,8). The fact that the diagnosis of thyroid cancer anteceded the diagnosis of FAP is consistent with a report from Donnellan et al. (4) who noted that TC may antecede colon cancer by 4–12 years, and a review by Cetta et al. (9) who found that one third of cases of CMVPTC were detected prior to the diagnosis of FAP. While this variant of PTC can be found sporadically, the majority of cases are associated with FAP, and diagnosis of this PTC variant should always be followed by a high clinical suspicion for associated FAP. Ultrasonography is not helpful because the CMVPTC lesions most often do not exhibit worrisome ultrasonographic features (10). As illustrated in this case, the differentiation of CMVPTC from aggressive PTC variants, such as tall-cell, columnar cell, and others (poorly differentiated, medullary, or hyalinizing trabecular tumors) can be challenging (5,11,12). Making the correct diagnosis is crucial for two reasons: 1) to potentially link the case to the FAP syndrome, and 2) for prognosis. Despite its peculiar morphology, CMVPTC is reported to have an excellent prognosis, similar to the classic variant of PTC (4). Interestingly, our patient exhibited local invasion at presentation as well as local recurrence or residual disease. While this is atypical, there have been other reports of aggressively behaving CMVPTC in the literature (5,12,13). Unfortunately, in our case, due to a lack of complete initial surgical records and initial postoperative surveillance, we are unable to distinguish between local recurrence and persistent disease. Ultimately, a tumor's biologic behavior may be closely associated with its specific molecular profile. As in our case, most CMVPTC associated with FAP is multicentric, while most sporadic cases present as single lesions (1).
Histology
Histologically, CMVPTC has a combination of epithelial and morular components. The epithelial component is arranged in a combination of papillary, trabecular, follicular, solid, spindle, and cribriform patterns (10,14) (Fig. 2A–2D). TTF-1 staining was positive (data not shown), while thyroglobulin staining was negative (Fig. 3D).The accumulation of β-catenin in the nucleus and cytoplasm represents the immunohistochemical hallmark of CMVPTC (6) (Fig. 3A). A second classic CMVPTC tumoral feature is the morule (Fig. 2B, arrow). Morules are “squamoid” areas with biotin-rich intranuclear inclusions responsible for the characteristic nuclear clearing (14) (Fig. 3C) seen in CMVPTC. This characteristic nuclear clearing is also seen in other low-grade malignancies associated with APC/β-catenin pathway dysregulation (11).
Molecular
The molecular alterations associated with CMVPTC are incompletely understood but seem to involve the WNT signaling pathway (1). Interestingly, to date, RET/PTC-1 is the only mutation classically associated with well-differentiated TC to be reported in TC cases associated with FAP. The molecular mechanisms implicated in FAP-associated CMVPTC are depicted in Figure 4. In 60%–80% of FAP cases, a germline mutation in the adenomatous polyposis coli (APC) tumor suppressor gene can be detected (1,11). The APC gene product is instrumental in the degradation and therefore the regulation of β-catenin (1). In the unstimulated state, APC is part of a destruction complex that binds to phosphorylated β-catenin and stimulates its proteosomal degradation. When the WNT pathway is stimulated, this destruction complex disassociates, and β-catenin is translocated to the cell nucleus where it activates gene transcription. The resulting gene products are involved in cellular proliferation, differentiation, migration, and apoptosis (15,16). The presence of an inactivating APC mutation resembles the stimulated state, which results in β-catenin accumulation in the cytoplasm and nucleus and unregulated activation of the WNT pathway (1). The location of the germline mutation in the APC gene appears to be important in establishing some of the phenotypic features of the FAP syndrome (genotype/phenotype correlation) (6,17). Germline mutations occurring before codon 1220 have a particularly strong correlation (p≤0.05) with the development of TC (7).
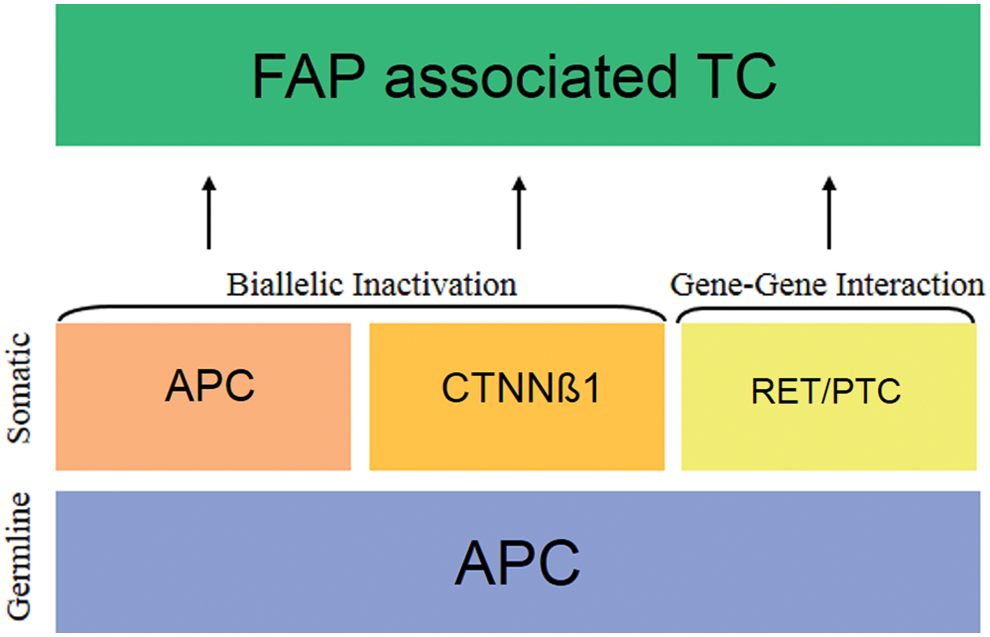
Proposed molecular mechanisms implicated in FAP-associated thyroid cancer. A germline adenomatous polyposis coli mutation is almost always detected. For the initiation of oncogenesis one or more additional molecular alterations must occur: new somatic mutations in the APC gene (biallelic inactivation), somatic mutations in the β-catenin gene (CTNNB1) (a “phenotypically equivalent” biallelic inactivation) or gene-gene interaction (epistasis) with other genes. Multiple mechanisms may coexist in the same patient (polygenic phenomenon). APC, adenomatous polyposis coli; CTNNB1, catenin (cadherin-associated protein), beta 1; RET/PTC, ret proto-oncogene/papillary thyroid cancer rearrangement; FAP, familial adenomatous polyposis; TC, thyroid cancer. Color images available online at
Although the inactivation of one of the ACP alleles by the germline mutation is considered the initiating event in FAP, it should not suffice on its own to cause significant dysregulation of the WNT pathway. This is because the remaining wild-type APC allele would be expected to compensate for the loss of function (7). How then does FAP result in the development of multiple tumors? In the case of CMVPTC, there seem to be at least three putative mechanisms (biallelic inactivation of the APC gene, phenotypic-equivalent mutations, and gene-gene interaction) as will be outlined in the following section. An alternative explanation is that multiple mutations may coexist, even in the same patient. The presence of distinct clonal origins is well established for the multicentric colon cancer lesions in FAP. Each individual colonic tumor can potentially harbor a different molecular signature (18,19). The same may be true for FAP-associated TC, which is also frequently multicentric (18,20).
For many FAP-associated tumors, such as colon cancer, inactivation of the second wild-type APC allele (“biallelic inactivation”) is necessary for tumorigenesis and occurs early in the carcinogenic process (14). The inactivation of the second APC allele is thought to occur most commonly as a result of a newly acquired somatic mutation. This new mutation can be located in any of the 15 exons of the APC gene, but is most often found in the mutational cluster region (MCR), a “hot spot” for mutations spanning codons 1285 to 1513 (20). Other mechanisms, such as methylation of the second APC allele, can also result in loss of heterozygosity. This rather common event for other FAP-associated malignancies has been documented in some, but not all cases of TC associated with FAP (21). Iwama et al. (21) were the first to report coexisting germline and somatic mutations in the TC tissue of two FAP patients. Miyaki et al. (19) reported two more cases of multicentric TC with not only coexisting germline and somatic mutations of the APC gene, but different somatic mutations (distinct clonal origins) in each tumor. Other authors have reported similar results (20). In contrast, others have extensively documented the absence of APC somatic mutations in FAP-associated TC, even outside the MCR (7,14,22,23). In summary, biallelic inactivation has not been consistently reported in FAP-associated TC. Of note, APC mutations have not been reported in the nonsyndromic, classic, well-differentiated form of PTC.
An alternative mechanism for inactivation of the second APC allele is the “phenotypic-equivalent” of biallelic inactivation, which has been reported once in both FAP-associated TC and sporadic CMVPTC (11,14). Somatic mutations in exon 3 of the β-catenin (CTNNB1) gene inhibit phosphorylation of β-catenin. The destruction complex cannot degrade β-catenin in the unphosphorylated state. As a result, β-catenin accumulates, translocates to the nucleus, and results in unregulated activation of the WNT pathway, similar to when inactivating APC mutations are present (1,17). Xu et al. (14) reported one case of multicentric FAP-associated TC with different CTNNB1 gene mutations in each tumor (distinct clonal origins). Germline CTNNB1 mutations have not been reported.
A third explanation is that a single mutated APC allele may suffice on its own in certain tissues (tissue-specific dominant effect) (7). This may be possible if other mechanisms (17) (environmental factors, hormones, etc.) or a second mutation “collaborates” with the APC germline mutation (gene–gene interaction) (23). Examples of this tissue-specific process are FAP-associated liver cancers, in which APC with p53, and colon cancer, in which the APC gene interacts with p53 and RAS (9). In the case of TC, the coexisting mutation uniformly reported is the RET/PTC-1 translocation (4,9,22,23). RET/PTC-1 has been reported in 80%–100% of TC cases associated with FAP (22,23). It has been postulated that APC may specifically predispose to the RET/PTC-1 translocation (23) since, untilnow, the other mutually exclusive mutations commonly found in PTC (BRAF, RAS, PAX/PPRγ) (11,24) have not been reported to coexist with APC (5,12,13). The exact mechanism by which APC and RET/PTC-1 may interact is unknown (23), and coexistence certainly does not necessarily imply interaction.
Conclusion
From a clinical and histologic perspective, this case represents an atypically aggressive presentation of CMVPTC associated with FAP.
The molecular alterations and mechanism responsible for the development of CMVPTC, are not well understood. Roughly 150 cases have been studied and reported. To date, only the RET/PTC-1 mutation has been reported in CMVPTC. To the best of our knowledge, this case represents the first published report of a RAS mutation in a case of CMVPTC. RAS is an upstream participant in the MAPK signal transduction pathway, and mutations of this protein have been documented in several cancers including non–small cell lung carcinoma (25) and colorectal carcinoma (26). The mutation discovered in this case, K-RAS Q61K represents a substitution at codon 61 of glutamine to lysine. The same mutation has been reported recently in lung (27,28), colorectal (29), pancreatic (30), and medullary thyroid cancer (31,32). Moura et al. (31) studied 25 samples of RET-negative MTC and found that 68% had a RAS mutation, including one with the Q61K mutation.
How this RAS mutation may participate in the molecular pathogenesis of CMVPTC is unknown. Given the more aggressive presentation and behavior of our patient's tumor compared to that classically described for CMVPTC, it is conceivable, but speculative, that RAS mutations may confer a worse prognosis than the RET/PTC-1 translocation.
More investigation is necessary to better understand the molecular mechanisms responsible for the development of this primarily FAP-associated and histologically discrete variant of PTC.
Footnotes
Author Disclosure Statement
No competing financial interests exist.