
Abstract
Background:
RET/PTC rearrangement, RAS, and BRAF mutations are considered to be mutually exclusive in papillary thyroid carcinoma (PTC). However, although concomitant mutations of RET/PTC, RAS, or BRAF have been reported recently, their significance for tumor progression and survival remains unclear. We sought to examine the prognostic value of concomitant mutations in PTC.
Methods:
We investigated 88 PTC for concomitant mutations. Mutation in BRAF exon 15, KRAS, NRAS, and HRAS were studied by polymerase chain reaction (PCR)-sequencing of tumor DNA; RET/PTC rearrangement was determined by reverse transcription (RT)-PCR-sequencing of tumor cDNA.
Results:
BRAF V600E was detected in 39 of 82 classic PTC (CPTC) and in all three tall-cell variants (49%, 42/85). KRAS mutation (p.Q61R and p.S65N) was detected in two CPTC (2%, 2/88) and NRAS Q61R in one CPTC and two follicular variant PTC (FVPTC; 3%, 3/88). KRASS65N was identified for the first time in thyroid cancer and could activate mitogen-associated protein kinase (MAPK). RET/PTC-1 was detected in nine CPTC, one tall-cell variant, and two FVPTC. Concomitant BRAF V600E and KRAS, or BRAF V600E and RET/PTC-1 mutations were found in two CPTC, and six CPTC and one tall-cell variant, respectively. In total, 11 concomitant mutations were found in 88 PTC samples (13%), and most of them were in the advanced stage of disease (8/11, 73%; p<0.01).
Conclusions:
Our data show that concomitant mutations are a frequent event in advanced PTC and are associated with poor prognosis. The concomitant mutations may represent intratumor heterogeneity and could exert a gene dosage effect to promote disease progression. KRASS65N can constitutively activate the MAPK pathway.
Introduction
T
It is generally believed that genetic alterations in RET/PTC, RAS, and BRAF are mutually exclusive in PTC, and mutations at more than one of these genes are unlikely to provide an additional biological advantage (2,5,6). However, recent studies have shown concomitant mutations in PTC (7 –10). Di Cristofaro et al. reported concomitant RAS and RET/PTC1 mutations in 1/24 (4%) follicular variant PTC (FVPTC), and BRAF and RET/PTC3 mutations in 1/26 (4%) classic PTC (CPTC) (8). Costa et al. showed concomitant BRAF and KRAS mutations in 4/35 PTC (11%) (10), whereas Henderson et al. reported concomitant BRAF and RET/PTC mutations in 5/54 (9.3%) recurrent PTC (7). Zhu et al. reported concomitant mutations of RET/PTC and RAS or BRAF in 8/15 (53%) subclonal or nonclonal PTC but none in clonal PTC (9). It is not clear whether concomitant mutations are associated with aggressive disease behavior and poor prognosis. In the present study, we investigated concomitant mutations in RET/PTC, RAS, and BRAF in 88 PTC from Saudi Arabia and its association with disease progression and prognosis.
Materials and Methods
Thyroid tumor specimens and cell lines
All tumor tissues were obtained at surgery with informed consent, and were immediately frozen in liquid nitrogen and stored at −70°C until processed. The clinical staging of thyroid cancer was based on the Tumor, Node, Metastasis (TNM) classification (11). Eighty-eight PTC diagnosed between 1987 and 2006 were selected randomly and included in the study: 82 CPTC, three tall-cell variants, and three FVPTC. The mean follow-up interval was 12 years. The following cell lines were studied to determine whether they have dual mutations in RET/PTC, RAS, or BRAF: K1, BCPAP, 8305C, 8505C, HTC-C3, BHT101, CAL62, TPC-1, and FRO. TPC-1 and FRO were kindly provided by Dr. James Fagin (Department of Medicine, Memorial Sloan-Kettering Cancer Center, New York, NY). Other cell lines were purchased from DSMZ (Braunschweig, Germany). ATC cell lines such as 8305C, 8505C, BHT101, FRO, and CAL62 were screened for concomitant mutations, since all of them carry either BRAF or RAS mutations, and it is not clear whether they have dual mutations in the MAPK pathway. The study was reviewed and approved by the Institutional Review Board.
Detection of BRAF and RAS mutations
Tumor tissue was obtained by standard sectioning, and DNA was extracted by standard proteinase-K treatment followed by phenol/chloroform extraction. BRAF exon 15 was amplified by PCR and directly sequenced as described previously (12). Mutations of KRAS, NRAS, and HRAS were analyzed by polymerase chain reaction (PCR) sequence analysis of exon 2 (for codon 12/13), exon 3 (for codon 61), and exon 4 in KRAS and NRAS; and exon 1 (codon 12/13), exon 2 (codon 61) and exon 3 in HRAS. Exon 4 was screened for non-hotspot mutations. PCR primers are listed in Supplementary Table S1 (Supplementary Data are available online at
Detection of RET/PTC rearrangements
Total RNA was extracted from tumor specimens and cell lines by the guanidine thiocyanate-phenol-chloroform method. The integrity of RNA was verified by denaturing gel electrophoresis. Two μg of total RNA were reverse-transcribed into cDNA using the Promega reverse transcription (RT) system (Promega, Madison, WI). Nested RT-PCR was used to amplify transcripts of RET/PTC rearrangements, using two sets of primers listed in Supplementary Table S1 and PCR conditions described above. The resulting PCR products were analyzed by gel electrophoresis and directly sequenced. GAPDH cDNA was used as an internal control for RNA quality. To rule out PCR contamination, positive samples were confirmed by repeating PCR using different batches of same specimen.
Screening for PAX8/PPARγ rearrangement in FVPTC
A fusion gene (PAX8/PPARγ) between the DNA binding domain of the thyroid-restricted transcription factor paired box gene 8 (PAX8) and the peroxisome proliferator-activated receptor γ gene (PPARγ) has been detected in about 10% of FVPTC. PAX8/PPARγ rearrangement was screened by RT-PCR in FVPTC as described previously (14).
Cloning and expression of mutant KRAS S65N and NRAS S65C
The full-length KRAS and NRAS cDNA clones were obtained from GenScript (Piscataway, NJ) and cloned into pcDNA3.1 under the control of the CMV promoter (Invitrogen, Carlsbad, CA). KRAS S65N and NRAS S65C mutants were obtained by site-directed mutagenesis and were verified by DNA sequencing. Wild-type KRAS and NRAS were used as controls. Equal amounts of the constructs were transfected into HEK293, BCPAP, and CAL62 cell lines using Lipofectamine (Invitrogen). Cells were collected 48 hours after transfection for MAPK and cAMP response element binding protein (CREB) activity.
Western blot analysis
Forty μg of protein from transfected cells was loaded onto a 12% SDS-polyacrylamide gel. Proteins were transferred to a polyvinylidene (PVDF) membrane and subject to Western blot analysis using anti-phospho-MEK1/2, anti-phospho-ERK 1/2 antibody, or anti-phospho-CREB (1:1000; Cell Signaling Technology, Inc., Danvers, MA).
Cell proliferation assay
Cell proliferation was measured by a non-radioactive MTS assay kit according to the manufacturer's procedure (Promega). Briefly, cells were plated in triplicate into 96-well plates (5×103 cells/well) containing 1% or 10% serum respectively for 48 h. At the final 4 h of incubation, 20 μL of CellTiter 96® AQueous One Solution reagent was added into each well for measurement of cell viability.
Statistical analysis
Fisher's exact test (two-tailed) was used. A p-value of ≤0.05 was considered significant.
Results
BRAF mutation
BRAF V600E mutation was detected in 39 of 82 classic PTC (48%) and in all three tall-cell variants (Fig. 1 and Table 1). No mutation was detected in FVPTC. The BRAF V600E mutation occurred more frequently in the advanced stages (stage III or IV) compared to the early stages (stage I or II) of CPTC: 17/26 (65%) versus 22/56 (39%) (Table 1; p<0.05, Fisher's exact test). The BRAF V600E mutation was also detected in 8305C, 8505C, HTC-C3, BHT101, BCPAP, K1, and FRO cells.
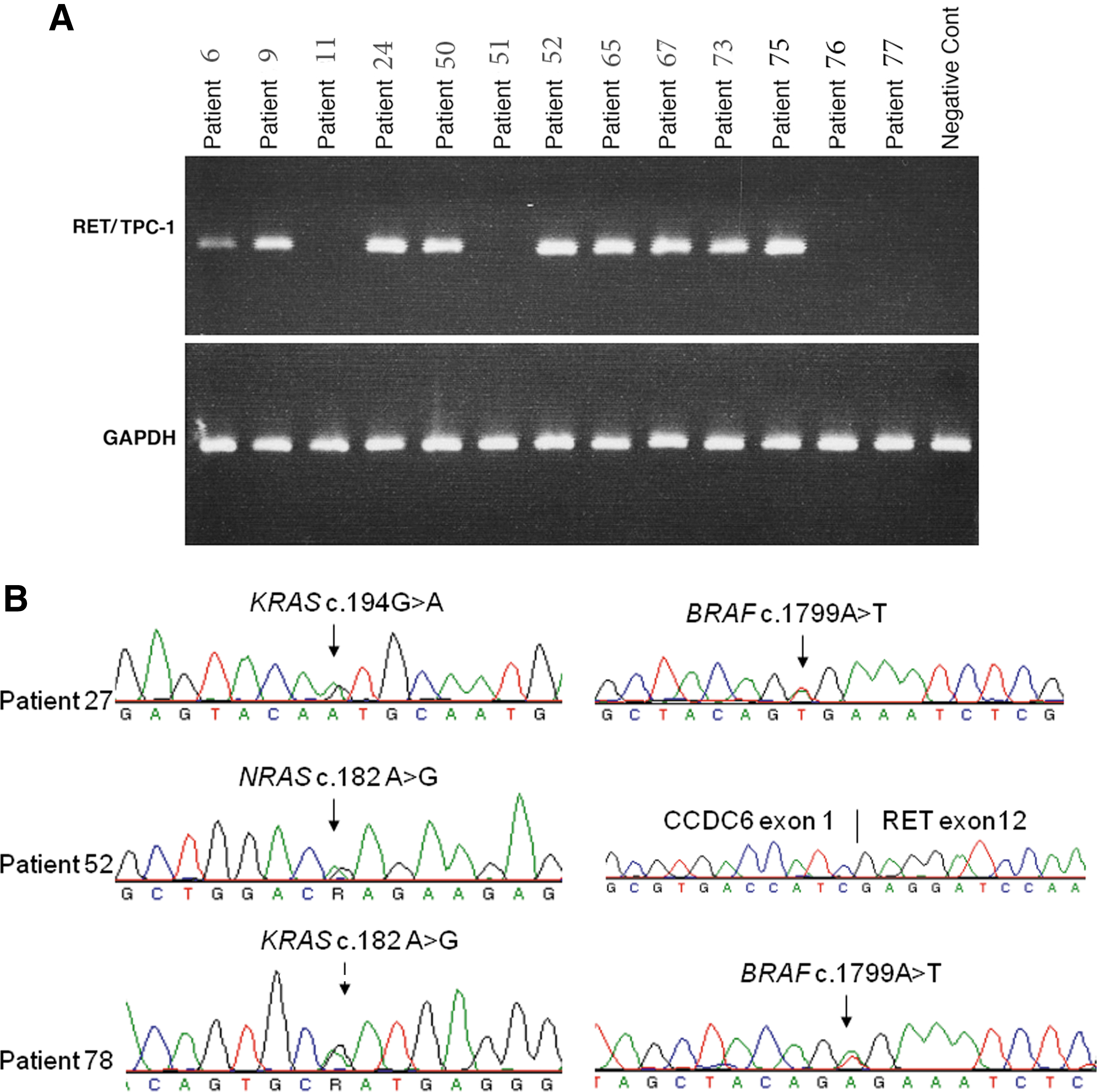
Detection of BRAF, RAS, and RET/PTC-1 mutations in papillary thyroid carcinoma (PTC).
MET PTC refers to stage I or II tumors with local lymph node metastasis. Dual mutations are highlighted in bold.
PTC, papillary thyroid carcinoma; ND, not detected.
RAS mutation
KRAS mutation was detected in two CPTC: c.182A>G (p.Q61R) with stage III cancer, and c.194 G>A (p.S65N) with stage I cancer (Fig. 1). NRAS
Q61R was detected in one CPTC case and two FVPTC cases. No mutation was found in HRAS. The overall rate of RAS mutation was 6% (5/88) in the cohort studied (Table 2). KRAS
G12R was found in the CAL62 cell line. KRAS
S65N has not been reported before in thyroid cancer. The significance of the mutation was further investigated first in matched normal tissue from paraffin blocks. No mutation was found in normal tissue, suggesting that it is not a germ-line polymorphism (data not shown). Multiple protein sequence alignments around codon 65 showed that serine65 is highly conserved across different species (data not shown). Next, we used PolyPhen-2 to predict the possible effect of S65N substitution (

Characterization of KRASS65N
and NRASS65C
on mitogen-associated protein kinase (MAPK) activity.
Numbers in parentheses indicate percentage of mutation in the histology subtype or group. HRAS mutation was not detected in the series.
RET/PTC rearrangements
A RET/PTC-1 rearrangement was detected in nine CPTC (9/82, 11%), one tall-cell variant, two FVPTC, and the TPC-1 cell line (Fig. 1 and Table 2). The overall rate of RET/PTC-1 was 14% (12/88). RET/PTC-2 and -3 were not detected in this series of samples. RET/PTC-1 occurred more frequently in the late stage (7/29, 24%) than in the early stage of PTC (5/59, 9%; p=0.0548).
PAX8/PPARγ rearrangement
No rearrangement was detected in the three FVPTC cases (data not shown).
Concomitant BRAF V600E , RAS, or RET/PTC-1 mutations
Two CPTC samples had both BRAF V600E and KRAS Q61R or KRAS S65N. Six CPTC and one tall-cell variant contained both BRAF V600E and RET/PTC-1. Two FVPTC samples had both NRAS Q61R and RET/PTC-1 (Table 1). None of the samples were found to have triple mutations. In total, concomitant mutations were found in 11/88 PTC samples (13%). Among them, three were found in early stages of PTC (3/59, 5%), and eight (8/29, 28%) in an advanced stage. The association of concomitant mutations with advanced stage was statistically significant (p<0.01). Eight cases with concomitant mutations were older than 45 years. Concomitant mutations were not found in nine cell lines studied. Co-existence of RET/PTC with RAS or BRAF has been reported in multiclonal PTC (9). We next examined different tumor sections of paraffin blocks for concomitant BRAF and RAS mutations found in our two cases. As shown in Table 3, the BRAF mutation was present in all tumor sections, whereas the RAS mutation was detected only in some sections. This may indicate intratumor heterogeneity. Histologic heterogeneity was not observed. Due to the limited sensitivity of Sanger sequencing to detect a low frequency of mutant alleles, we cloned the PCR fragments from tissue sections with negative RAS mutation into a TA vector, and screened 50 clones from each fragment for RAS mutation by direct Sanger sequencing. As shown in Table 3, a RAS mutation was detected in two sections that were negative by direct Sanger sequencing. We further evaluated the sensitivity of Sanger sequencing to detect KRAS mutations by mixing different amounts of wild-type and mutant KRAS DNA. As shown in Figure 3, about 20% mutant alleles are required in the sample to be reliably detected by Sanger sequencing (the detection limit is about 20%). In the lymph node metastases, the RAS mutation was significantly enriched and can be easily detected (Table 3).

Sensitivity of Sanger sequencing to detect KRAS mutations. Different amounts of wild-type and mutant KRAS DNA were mixed together in a total concentration of 100 ng DNA for PCR. PCR products were directly sequenced. At least 20% mutant alleles need to be present in the sample to be reliably detected by Sanger sequencing.
PT, primary tumor; LNM, lymph node metastasis; NT, not detected by direct Sanger sequencing.
To determine whether concomitant mutations were associated with a poor clinical outcome, we analyzed disease-free survival of the patients who had concomitant mutations versus those without mutation or those who had only one mutation. As shown in Figure 4, the Kaplan–Meier curve shows a significant difference in the recurrence-free probability over a 10-year period among patients with no mutation, a single mutation, and double mutations (log-rank test χ2=18.2, p<0.0001), and between patients with single and double mutations (log-rank test χ2=4.2, p<0.041). These data indicate that patients with concomitant mutations had much poorer clinical outcomes than those with a single or no mutation.
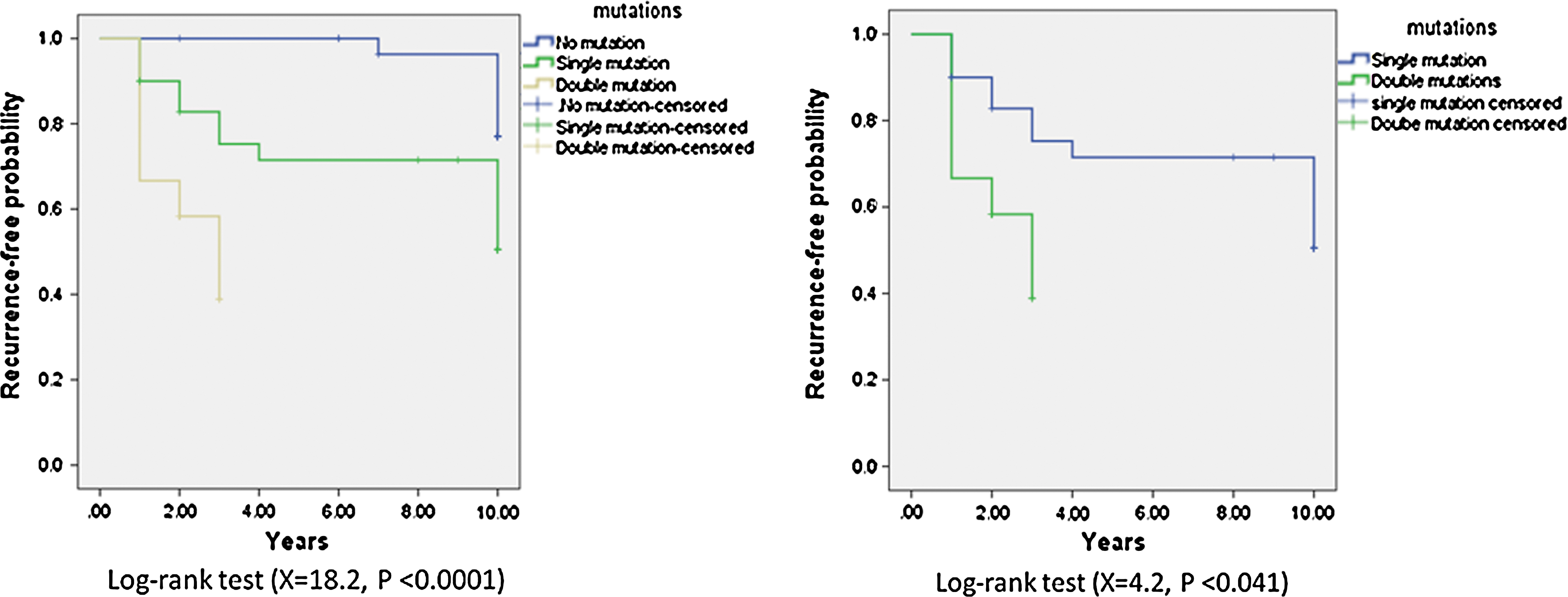
Kaplan–Meier analysis for recurrence-free probability. The Kaplan–Meier curves shows a significant difference in the recurrence-free probability over a 10-year period after the diagnosis among patients with no mutation, a single mutation, and double mutations (log-rank test χ2=18.2, p<0.0001), and also between patients with single mutation and double mutations (log-rank test χ2=4.2, p<0.041).
Discussion
In the present study, we found 11 cases of PTC (13%) with concomitant BRAF, RAS, or RET/PTC-1 mutations. Concomitant mutations occurred more frequently in advanced stages of disease. Long-term follow-up showed that patients with concomitant mutations had a poor response to treatment and shorter disease-free survival.
We also report the first case of KRASS 65N in PTC (1%). Mutation in KRAS codon 65 has been reported in one case of cervical cancer (1/107, 1%) (15). A codon 65 mutation in NRAS has been reported in 2/12 (17%) neuroendocrine lung cancers and 1/35 (3%) melanomas (16). The melanoma with NRASS 65C also had BRAF V600E (16). The significance of codon 65 mutations in RAS has not been investigated previously, although the residue is highly conserved across different species. We demonstrate in the present study that codon 65 mutations in both KRAS and NRAS can constitutively activate the MAPK pathway. To our knowledge, this is the first report showing the functional significance of a RAS codon 65 mutation. The prevalence of a codon 65 mutation appears to be low in human cancers. However, many studies have only screened RAS mutations at codon 12/13 and 61, and may miss this mutation. Two of three FVPTC (66.7%) in our study have NRAS mutations. A NRAS mutation is more frequent in FTC (17) and FVPTC (8), supporting the notion that FVPTC shares many molecular features of FTC.
The overall rate of RAS mutation is 6% (5/88) in the cohort studied. The frequency of RAS mutation in PTC is variable depending on the cohort studied, ranging from 0% to 16% (2,8,18), with an overall rate of 11% (17). Higher frequencies have been reported in poorly differentiated thyroid carcinomas (23%) (19) and FTC (20%) (17). In the study by Wang et al. (18), a RAS mutation was found in 3 of 31 FTC (10%) and none of the 141 PTC in a Chinese population. Similar results were also reported by Di Cristofaro et al. in a French population (8). In contrast, Kimura et al. reported a RAS mutation in 11 of 67 PTC (16%). As discussed by Vasco et al. (17), multiple factors may contribute to such a wide range of variations, including the methodology used to detect mutations (the overall rate of mutations was significantly lower when estimated with direct sequencing than without: 12.3% vs. 17%), environmental factors (iodine deficiency, radiation exposure, and food-borne carcinogens), geographic variation, and tumor heterogeneity. Based on the pooled literature analysis by Vasco et al. (17), the most common mutation in PTC is NRAS Q61R (5%), whereas the KRAS and HRAS mutation rate is about 3% and 2.6% respectively.
Concomitant RAS and BRAF mutations have been reported previously in PTC, poorly differentiated and anaplastic thyroid carcinomas (10), colorectal cancers (20), and cell lines (21). By using pyrosequencing to quantitate BRAF and RAS mutations, Guerra et al. demonstrated simultaneous subclonal BRAF and RAS mutations in two PTC (3%) (22).The frequency of concomitant KRAS and BRAF mutations increased along progression of colorectal cancers, suggesting that activation of both genes may have a synergistic effect for disease progression (20). In our two cases with concomitant RAS and BRAF mutations (one of them was a stage I PTC at diagnosis), they responded poorly to treatment. The disease relapsed within five years after surgery and progressed aggressively. Given that the BRAF mutation is present in all tumor sections and the RAS in some sections, it is likely that the RAS mutation occurred later than the BRAF and may play a role in disease progression. In the four cases of concomitant RAS and BRAF mutations reported by Costa et al. (10), two cases show a negative RAS and a positive BRAF mutation in some primary tumor sections, but concomitant RAS and BRAF mutations are present in all sections of lymph node metastasis, supporting the role of RAS in disease progression. RAS can promote thyroid tumorigenesis through both the MAKP and PIK3CA/AKT pathways. Concomitant RAS and BRAF mutations may allow simultaneous activation of both signaling pathways in cancer cells to gain growth advantage. Indeed, activation of AKT has frequently been found in PTC and is associated with cancer progression (23,24). PIK3CA and AKT mutations also frequently coexist with RAS and BRAF mutations in patients with advanced cancers (10,25,26). Although occurring less frequently, AKT1, AKT3, and PIK3CA mutations have been reported in PTC (27,28).
BRAF inhibitors have been undergoing clinical trials for thyroid cancer with mixed results (29,30). Recent studies have demonstrated that BRAF inhibitors may paradoxically increase MAPK activation in the presence of mutated RAS or wild-type BRAF (31 –33). Therefore, patients with concomitant RAS and BRAF mutations may not be suitable for BRAF inhibitor treatment. Reactivation of MAPK or activation of PIK3CA/AKT (34) plays an important role in BRAF inhibitor resistance (35). MAPK reactivation can occur via NRAS mutations, COT overexpression, BRAFV600E alternative splicing or amplification, and MEK1 mutation, whereas PIK3CA/AKT can be activated through overexpression of receptor tyrosine kinases (35). RET/PTC can stimulate both PIK3CA/AKT and MAPK pathways (6,36,37). For patients with concomitant RET/PTC and BRAF mutations, it remains to be determined whether RET/PTC plays any role in BRAF inhibitor resistance, since cancer cells may escape inhibition via PIK3CA/AKT survival pathway. Targeting both MAPK and PIK3CA/AKT pathways may offer better long-term therapeutic effects. Selection of patients based on their mutation profile for clinical trials or treatment strategies may be warranted.
RET/PTC-1 was detected in 12 cases of PTC (14%). The presence of a RET/PTC rearrangement is not an indication of malignancy, as it occurs in benign thyroid nodules and is associated with a high growth rate of these nodules (38). Papillary carcinomas are often multifocal, and individual tumor foci in patients with multifocal PTC often arise from independent clonal origins (39). Based on the methodology used in the current study, we cannot distinguish whether dual mutations are present in the same cell or in different cells of the same tumor. We found concomitant mutations of RET/PTC-1 and BRAF or RAS in nine cases of PTC but none in the studied cell lines, suggesting intratumor heterogeneity or multiclonal origin in tumor samples and monoclonal origin of cell lines. The mutual exclusivity of gene mutations in the MAPK pathway may still hold if tumors are of monoclonal origin. A recent study of a single kidney cancer has shown remarkable diversity in the genetic changes that have taken place in different parts of the tumor (40). In advanced thyroid carcinoma, multiple genetic changes may occur in different parts of the tumor, resulting in concomitant mutations. Among nine cases with concomitant RET/PTC-1 and BRAF or RAS mutations, seven cases (78%) are in the late stage of cancer, indicating that RET/PTC-1 may occur late in subclonal PTC and is involved in tumor progression, as suggested previously by Unger et al. (41). Recently, Guerra et al. also reported concomitant subclonal BRAF and RET/PTC mutations in 19.4% (14/72) case of PTC (42). RAS mutations may have been underestimated in thyroid cancer. Using sensitive allele-specific competitive blocker-PCR (ACB-PCR), which can detect less than 1% mutant alleles, Myers et al. detected KRAS mutations in 30% of PTC (43). Although Sanger sequencing is less sensitive than pyrosequencing (detection limit is about 5%) or ACB-PCR to detect RAS mutations, mutations detected by Sanger sequencing indicate that at least 20% mutant alleles are present in the tumor sample, which may play a more significant role in promoting tumor progression. The oncogenic effects of RET/PTC-1 are mediated via RAS, which allows growth promoting signals through both the MAPK and PIK3CA/AKT pathways, and concomitant mutations of RET/PTC-1 and BRAF or RET/PTC-1 and RAS in subclonal/multiclonal PTC may provide a synergistic effect for disease progression.
Footnotes
Acknowledgment
The study is supported by a Biotechnology grant #10-BIO957-20 from King Abdulaziz City for Science and Technology.
Author Disclosure Statement
All authors have nothing to declare.