
Abstract
Background:
Age-related risk of medullary thyroid carcinoma (MTC) development in presymptomatic carriers of lower risk germline RET mutations is uncertain; such data may aid counseling patients regarding timing of thyroidectomy.
Methods:
From an institutional database and an exhaustive literature review, we identified 679 patients with American Thyroid Association (ATA) level A or B mutations who were identified because of family screening (index cases of MTC were excluded to minimize selection bias). We evaluated age at thyroidectomy or last evaluation if no thyroidectomy, preoperative calcitonin level (elevated or not), the mutated codon, and outcome (MTC vs. no MTC after thyroidectomy or no clinical evidence of MTC if thyroid intact). Data were used to estimate the cumulative prevalence of MTC and/or assess likelihood of MTC stratified by codon. After exclusion of cases with missing data or small representation, 503 patients with mutations in codons 533, 609, 611, 618, 620, 791, and 804 were analyzed.
Results:
236 patients had MTC. Cumulative prevalence and median time to MTC varied by codon and within ATA risk levels (p<0.0001). Patients with a codon 620 mutation were 2.8–6.9 times more likely to have MTC than other level B mutation carriers, and 5.1–21.7 times more likely than level A mutation carriers included in our focus population. The youngest median time to MTC was 19 years for codon 620 and the oldest was 56 years for codon 611. Cumulative prevalence of MTC by age 20 was 10% or lower for codons 533, 609, 611, 791, and 804. By age 50, it ranged from 18% for codon 791 to 95% for codon 620. An elevated preoperative calcitonin level strongly predicted MTC on final pathology, though false-negative rates varied by codon (p<0.0001). Positive predictive values ranged from 76% to 100% by codon with an overall positive predictive value of 87% across codons.
Conclusions:
This study offers a better understanding of the age-related development of MTC in lower risk RET mutation carriers, provides evidence of further distinctions between lower risk mutations within ATA subgroups, and clarifies the clinical significance of codon 791 mutations. The data support individualized “codon-based” management approaches coupled with clinical data such as calcitonin levels.
Introduction
H
Because MTC develops in the vast majority of RET mutation carriers, early thyroidectomy is widely accepted as an effective method of preventing or curing MTC before it becomes metastatic. In 2001, genotype-based consensus recommendations stating that the particular RET codon mutation could be used to determine the timing of early thyroidectomy were published (5), and a second set of guidelines was issued in 2009 by the American Thyroid Association (ATA) (6). The ATA panel recommended classification of RET mutations into one of four categories based on risk and timing of development of MTC and aggressiveness of disease. ATA levels C and D are the highest risk levels, and mutation carriers are recommended to undergo thyroidectomy in early childhood. ATA levels A and B (ATA-A; ATA-B) include the “lower” risk RET mutations, in which the lifetime MTC risk is high but is typically of later onset and is less aggressive compared with level C and D carriers (7). In addition to early thyroidectomy, prospective clinical monitoring in ATA-A and ATA-B presymptomatic mutation carriers was introduced. As long as the diagnosis of MTC is not suggested through annual measurements of serum calcitonin and thyroid ultrasonography, thyroidectomy may be delayed based on patient and parent preference and with consideration for the aggressiveness of MTC in the family.
While this recommendation allows for added individualization of care, prospective data regarding the safety and efficacy of monitoring in these patients are not widely available and may not be broken down by individual codon mutations. Additionally, many studies include index cases of MTC, which may overestimate MTC risks because of selection bias. Until sufficient time has passed for such data to be generated, we chose to retrospectively review the mutation- and age-related prevalence of MTC in the ATA-A and ATA-B mutation carriers who present presymptomatically. Data that estimate the likelihood of MTC by age may aid in counseling presymptomatic patients regarding the timing of thyroidectomy, both in children and those genetically diagnosed as carriers when they are adults. Based on the growing body of literature regarding genotype–phenotype correlations, we hypothesized that further distinctions between low-risk (or ATA-A and ATA-B) RET mutations with regard to age-related progression of MTC could be determined.
Patients and Methods
Patients
Eligible patients were those with an ATA-A or ATA-B germline RET mutation who were screened for MTC only because of an identified RET mutation or family history of MTC or MEN2. All patients had a germline RET mutation, confirmed through genetic testing, or were obligate carriers whose mutation status could be definitively inferred from pedigree analysis. Index cases of MTC who were diagnosed after presenting symptomatically were excluded to avoid selection bias.
Patients were identified from two sources, a comprehensive review of published cases and a retrospective MEN2 database maintained at the University of Texas MD Anderson Cancer Center (MDACC). The primary literature search was conducted in March 2013 using PubMed and limited to articles on humans and published in English. Search terms included medullary, codon, and RET. References were also reviewed to identify additional eligible publications. Publications were excluded if they were review articles, were from MDACC, reported patients in aggregate only, or did not have sufficient information on age, mutation, ascertainment, and MTC status on individual patients. Publications from the same group were reviewed in detail to avoid duplicate patient records, and the most complete and up-to-date data were used preferentially.
Variables
Eligible patients were evaluated for the mutated codon, extent of surgery if performed, basal serum calcitonin levels (measured preoperatively or at last follow-up if no surgical intervention, recorded as elevated or not), and MTC status. MTC status was classified into one of the following four groups: (i) MTC (confirmed on pathologic evaluation), (ii) no evidence of MTC based on final pathologic evaluation after thyroidectomy, (iii) C-cell hyperplasia (CCH) on pathologic evaluation after thyroidectomy, and (iv) no evidence of MTC based on normal clinical evaluation (normal basal calcitonin level and/or thyroid ultrasonography) in patients who had not undergone thyroidectomy.
Patients who had MTC were further classified by lymph node status. Patients were considered to have no lymph node involvement only if their surgery included a lymph node dissection and none were involved with MTC or, for prior studies, if the authors reported TNM staging data and patients were specifically classified as N0. Age was recorded at the time of MTC diagnosis or, for patients who were not diagnosed with MTC, age was recorded at time of thyroidectomy or at last known follow-up if thyroidectomy was not performed.
Statistical analysis
Descriptive data (percent, median, range) were generated for the population overall as well as separately for each codon. Chi-square test or Fisher's exact test was used to evaluate the association between two categorical variables. The Kaplan–Meier method was used to estimate the cumulative prevalence of MTC and the median time to MTC, and the log-rank test was used to evaluate the difference among codons. Observation time was taken from birth to the age at pathologic diagnosis of MTC. For patients without pathologic or clinical evidence of MTC, observation time was taken from birth to age at thyroidectomy or last follow-up, respectively. Cox proportional hazards model was utilized to assess the hazard ratio of MTC between codons. Statistical analyses were performed using SAS 9.1.3 (SAS Institute Inc., Cary, NC) and S-Plus 8.0 (TIBCO Software Inc., Palo Alto, CA).
Results
Patient ascertainment
A total of 366 publications were identified and reviewed, from which 72 contained sufficient data on individual patients meeting eligibility requirements. We identified 601 patients with an ATA-A or ATA-B mutation who were not index cases, of whom 344 were from case/family reports and 257 from institutional experiences. An additional 78 were identified from the MDACC database for a total of 679 eligible patients.
RET codon mutations occurring at a frequency >2% within this population (n=581) included 804 (n=191), 618 (n=118), 609 (n=76), 533 (n=71), 791 (n=52), 620 (n=38), or 611 (n=35). The remaining patients (98 of the 679) had a mutation in one of 17 other codons: codon 891 (n=13), codons 631/790/649 (n=11 each), codons 912/666 (n=8 each), codon 630 (n=7), codon 768 (n=6), codons 321/600 (n=5 each), codon 531 (n=4), codons 603/635/777 (n=2 each), and codons 515/606/633 (n=1 each). Detailed data were collected and further analyzed only for the patient group with a higher mutation frequency (581 patients with mutation targeting 7 codons) (8 –58).
Of these 581, 78 cases (13%) had to be excluded because of missing data. These included 28 who were alive and well but not evaluated for MTC (median age of 49), 12 obligate carriers who died before being evaluated for MTC (median age of death of 76), 27 with hypercalcitoninemia without pathologic confirmation of MTC (excluded because of inability to determine CCH vs. MTC; median age of 50), and 11 whose MTC status and/or age was unclear (Fig. 1).
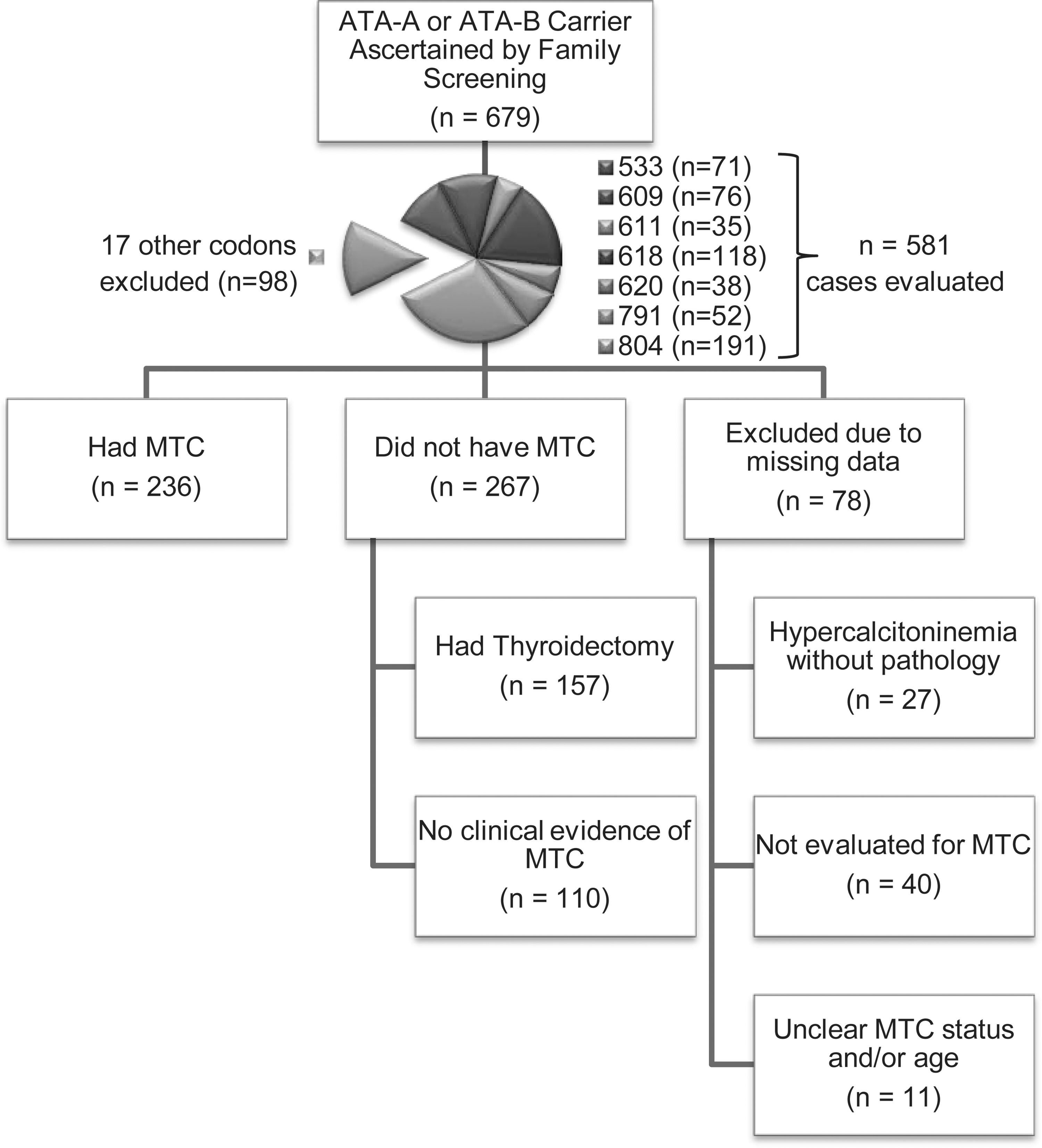
Study schema.
Overview of patient population and MTC status
Our final study population consisted of 503 mutation carriers (51% female) who were identified by family screening and represented 7 different ATA-A or ATA-B mutations (804, n=160; 618, n=109; 609, n=73; 533, n=49; 791, n=43; 620, n=38; or 611, n=31). Cumulative prevalence of MTC by age estimated by the Kaplan–Meier method did not differ between patients at MDACC and the cohort derived from the literature (p=0.204), and so the data sets were combined for further analyses. A detailed description of MTC status broken down by codon is provided in Table 1.
Sex not specified (n=14).
At last follow-up.
At surgery.
Lymph node status was defined as N0 only if the surgery included a lymph node dissection and none were involved with MTC or, for prior studies, if the authors reported TNM staging data and patients were specifically classified as N0.
At MTC diagnosis.
CCH, C-cell hyperplasia; MTC, medullary thyroid carcinoma; NED, no evidence of disease (MTC).
Overall, 236 patients (47%) of our final study population had MTC, though the proportion within each codon who had a diagnosis of MTC ranged from 14% for codon 791 to 74% for codon 533. All but one MTC patient had surgery; he is a codon 618 mutation carrier who was found with a 1.2 cm thyroid nodule with biopsy-confirmed MTC at MDACC at the age of 78 and declined surgery. At last follow-up at age 92, he is still alive and has had minimal MTC growth according to ultrasonography. Two hundred seven (88%) of the MTC patients who underwent surgery had data on regional and distal metastases at diagnosis. One hundred forty-four (70%) had no lymph node metastases and 62 (30%) had locoregional metastases. The median age at surgery was 38 (5–75 years) for patients with N0 MTC and 42 (10–87 years) for patients with N1 MTC (Table 1). Three patients with regional metastases also had distant metastases discovered at the time of their initial MTC diagnosis. Two are patients with a codon 618 mutation who had liver metastases at the time of diagnosis at age 27 and 26 (36); both are still alive with MTC 27 and 28 years later. Preoperative calcitonin levels were not reported. The third is an MDACC patient with a codon 620 mutation who had a preoperative calcitonin level of 11,800 pg/mL (normal was <17 pg/mL). She was initially diagnosed with MTC with bone and liver metastases at age 33. She received radiation to the bone metastases and was treated with systemic therapy on multiple clinical trials; she is still currently alive with MTC at age 43.
Of the remaining 267 patients who did not have MTC, 157 had a thyroidectomy with benign pathology, of whom 116 had CCH. The median age at thyroidectomy was 13 (range 2–65 years) for patients with normal thyroid gland and 19 (2–63 years) for those with CCH. The 110 patients with intact thyroid glands were evaluated clinically for MTC, and found to have normal calcitonin level and/or thyroid ultrasonography. The median age at last evaluation overall was 25 (range 1–86 years), but ranged from 5 years for codon 620 to 42 years for codons 609 and 791 (Table 1).
Preoperative calcitonin
Only 228 (58%) of the 392 surgical patients had data on preoperative basal calcitonin levels. MTC patients were more likely to have an elevated preoperative calcitonin level than patients with benign pathology (75% vs. 17%, p<0.0001). Fourteen of the 15 patients with elevated preoperative calcitonin and no MTC had CCH. Thirty-five of the 109 (32%) patients with a normal preoperative calcitonin had MTC (Table 2). The ability of an elevated calcitonin level to predict MTC on final pathology did not differ by codon. However, the false-negative rate did differ by codon (Table 2; p<0.0001). A detailed description of the patients with MTC with normal preoperative calcitonin levels is provided in Table 3. The median age of diagnosis of the patients with MTC and normal preoperative calcitonin levels was 34 (range 5–63 years); nearly all had microscopic MTC and all but two had N0 disease. The two patients with N1 disease harbored a RET codon 618 mutation and had T1 tumors (3 and 18 mm) with microscopic lymph node metastases (28). They were 27 and 48 years old at the time of surgery. Both had positive stimulated calcitonin levels; thyroid ultrasonography findings were not reported.
Positive predictive value refers to the number of patients with elevated CTN who had MTC divided by the total number of patients with elevated CTN.
Comparison of proportion with MTC by codon among patients with an elevated preoperative CTN.
False-negative rate refers to the number of patients with normal CTN who had MTC divided by the number of patients who had MTC.
Comparison of proportion of MTC by codon among patients with a normal preoperative CTN.
Overall comparison of MTC rates by whether CTN was elevated or not, p<0.0001.
ATA, American Thyroid Association; CTN, calcitonin.
Lymph node metastases measured <2 mm.
Cystic.
n/a, not applicable; NS, not specified; MDACC, MD Anderson Cancer Center.
Estimated cumulative risk and predictors of MTC
Figure 2 shows the estimated cumulative probability of MTC, and the corresponding table below shows the cumulative MTC prevalence by age with 95% confidence intervals. The Kaplan–Meier curve was similar when patients who did not undergo surgery were excluded from the analysis and were similar for males and females. Cumulative prevalence of MTC in children (by age 20) was 10% or lower for codons 533, 609, 611, 791, and 804. By age 50, cumulative prevalence of MTC ranged from 18% for codon 791 to 95% for codon 620. Patients with a codon 620 mutation had the youngest median time to MTC (19 years), followed by patients with mutations of codons 618 (28 years), 533 (47 years), 609 (50 years), 804 (54 years), and 611 (56 years). Median time to MTC could not be estimated for codon 791 as this endpoint was not reached. Overall, a difference in age-related cumulative prevalence of MTC existed by codon (p<0.0001). Patients with a codon 620 mutation were almost seven times as likely to have MTC than patients with a codon 611 mutation, even though both are grouped together as level B mutations (hazard ratio 6.97, 95% confidence interval [CI 3.59–13.54]; p<0.0001), and more than 21 times more likely to have MTC than codon 791 carriers (hazard ratio 21.71 [CI 8.66–54.42]; p<0.0001; Table 4).
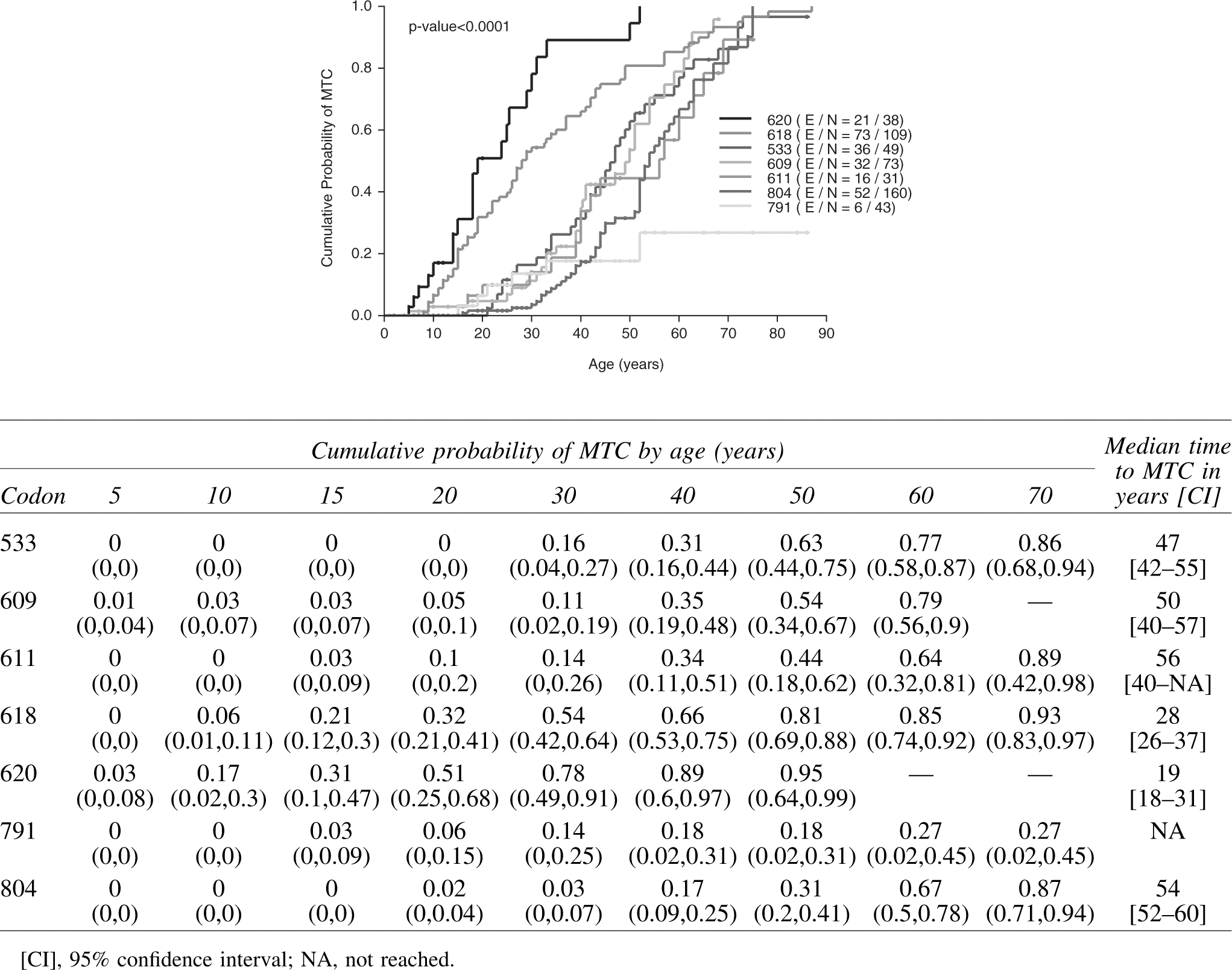
Cumulative probability of MTC in 503 ATA-A and ATA-B RET mutation carriers ascertained by family screening who underwent pathologic or clinical evaluation. Observation time was taken from birth to the age at pathologic diagnosis of MTC. For patients without pathologic or clinical evidence of MTC, observation time was taken from age at thyroidectomy and last follow-up, respectively. Cumulative probability of MTC by age and median time to MTC (95% confidence interval) is displayed in the table above. ATA-A, American Thyroid Association level A; ATA-B, American Thyroid Association level B; MTC, medullary thyroid carcinoma.
Because basal calcitonin levels are time-dependent and were not observed from birth in this study, this variable is not a baseline variable and as such could not be included in the Cox proportional hazards model. In order to control for calcitonin level in comparing MTC prevalence among codons, the Kaplan–Meier analysis was repeated in the 109 patients who had a thyroidectomy and a normal preoperative basal calcitonin level (Fig. 3) and in the 119 patients with elevated preoperative basal calcitonin levels (data not shown). A difference in cumulative prevalence of MTC among codons was still observed for both groups (p<0.05).
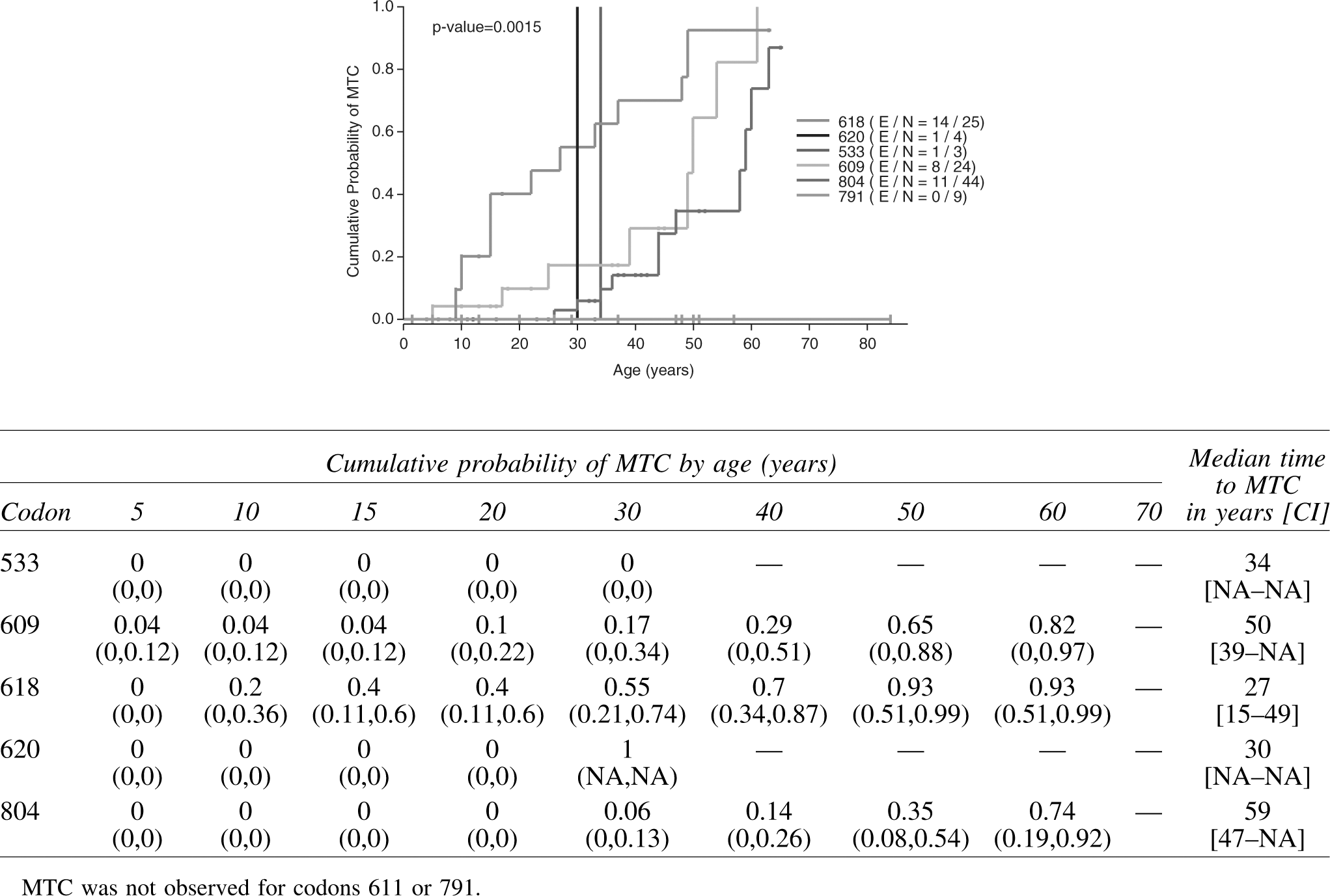
Cumulative probability of MTC in 109 ATA-A and ATA-B RET mutation carriers ascertained by family screening who had a normal preoperative basal calcitonin level. Observation time was taken from birth to the age at pathologic diagnosis of MTC. For patients without pathologic evidence of MTC, observation time was taken from age at thyroidectomy. Cumulative probability of MTC by age and median time to MTC [CI] is displayed in the table above.
Discussion
In the two decades since the discovery of RET germline mutations as the molecular etiology of MEN2A, guidelines have evolved to recommend RET testing in any patient diagnosed with MTC as well as in at-risk family members (6,59). Many of these individuals possess lower risk ATA level A or B mutations, which are often associated with a later age of onset of MTC and a less aggressive clinical course. The result of these efforts has been the emergence of a group of presymptomatic patients who are diagnosed with MEN2, often at a young age, yet have no clinical evidence of MTC. As more of these patients present for evaluation, additional information about this subgroup may be helpful to more effectively counsel patients about expectations of disease course and treatment and prevention options. To our knowledge, this study is the largest systematic series evaluating age-related prevalence and predictors of MTC focused specifically on lower risk asymptomatic RET mutation carriers.
The findings of this study support the presence of genotype–phenotype variability in presentation of MTC in the asymptomatic population, with differences found in cumulative probability of disease among codons with respect to age, as seen in the Kaplan–Meier curve (Fig. 2), and calcitonin level (Fig. 3). Of the level B mutations studied here, codon 609 and 611 mutations appear to have a later onset of MTC compared with carriers of codon 620 or 618 mutations, suggesting that perhaps the categorical division of codon mutations in this low-risk group of patients may not yet be optimized. For example, the age of onset curve of the level B 609 mutation appears more similar to that of the level A 804 mutation than it does to its fellow level B 620 mutation. Our data also confirm that lifetime risk of MTC is high for most codon mutations; however, the codon 791 mutation appears to form a unique category as the risk for MTC is much lower than that for the other codons.
The pathogenicity of codon 791 mutations has been debated given the many reports of codon 791 carriers who do not have clinical evidence of MTC at elderly ages, and codon 791 mutations have been detected in a similar proportion of MTC patients as population-based controls (0.9% vs. 0.8%), as well as in patients with sporadic lymphoma and brain and gastrointestinal carcinomas (60 –64). Our data on codon 791 carriers help clarify the clinical significance of this mutation. Only 6 of the 49 codon 791 carriers had MTC with an estimated cumulative probability of MTC of 27% by age 70. An additional 9 patients had CCH at a median age of 30 years (range 6–44 years). Interestingly, all 6 of the MTC patients with a codon 791 mutation in this study are part of the same kindred from Brazil (55). The index case did have sequencing of RET exons 8, 10, 11, and 13–16, though the relatives were tested only for the presence of the 791 mutation, making it possible but unlikely that another deleterious RET mutation was co-segregating with the 791 mutation. The observation of only a single family with multiple cases of MTC with this mutation suggests that there may be other modifying genes that must be present for MTC development and consequently that codon 791 mutations alone may not be pathogenic in many families.
It is notable that codon 533 mutations were the fourth most common mutation in the entire population of ATA-A and ATA-B mutation carriers ascertained by family screening, given that it is considered one of the rarer mutations. Codon 533 is located in exon 8 of the RET proto-oncogene, a region that is not routinely sequenced in most commercial laboratories in the United States offering RET analysis; therefore, patients with this mutation could easily be missed, which might account for its apparent rarity. Even though the majority of the 71 codon 533 patients came from a single large Brazilian family or from one of 5 different families of Greek ancestry (10,16,33,47), our data argue in favor of routinely sequencing exon 8, given that it is now a well-characterized and clearly deleterious mutation.
Our study confirms findings from others that calcitonin levels may be normal in patients with MTC (65,66). A novel finding in this study is the possibility that the negative predictive value of calcitonin may be lower for the “more aggressive” codons, perhaps because the a priori probability of MTC in these groups is higher. We observed that, in the setting of a normal calcitonin level, certain mutations continued to be associated with a younger age of onset and higher cumulative prevalence of MTC. Therefore, even within the low-risk categories, RET genotype should continue to factor into management decisions. Admittedly, this study's data on calcitonin are limited by having to be qualified as either “elevated” or “not elevated” because of changing assays and reference ranges over the time period of this study, in addition to how this variable is often reported in the literature.
We also acknowledge that the false-negative rate of calcitonin is a limitation in the cumulative prevalence of MTC estimates, in that we chose to include patients with normal calcitonin levels who did not undergo thyroid surgery and defined them as not having MTC, when they may in fact have had an occult MTC. Therefore, our data likely underestimate the true prevalence of MTC that one would observe on final pathology. However, in an effort to avoid introducing bias for patients who were selected for surgery, we chose to include them. Additionally, many of the surgical patients were young children undergoing early thyroidectomy. Therefore, if we had excluded older children and adults without clinical evidence of MTC who did not undergo surgery, we would have likely overestimated the true age-related prevalence.
What remains unknown is whether or not long-term outcomes of asymptomatic patients with lower risk RET mutations are improved by an earlier surgical intervention when the basal calcitonin level is still normal compared with those who have a slight to moderate increase. Because the patients included in this study were not followed after thyroidectomy in a systematic way, we were not able to analyze outcome data in this cohort even when follow-up data were available. Even for the MDACC cohort, patients with more significant disease are more likely to be followed at our center, which would significantly bias our findings. Even so, as illustrated in Tables 2 and 3, most patients with a normal calcitonin did not have MTC, and nearly all who did had microscopic MTC not associated with lymph node metastases. While the risk for persistent and recurrent MTC in patients with hereditary MTC remains most strongly associated with an elevated preoperative calcitonin level, risk of recurrence is low if the preoperative basal calcitonin is below 30 ng/mL (67). Additional studies may help clarify whether or not there is an identifiable value of calcitonin that defines clinically significant MTC in terms of disease-free or overall survival, and whether this value differs by codon.
Our data support findings from many other studies that onset of MTC, and particularly N1 MTC, is later for lower-risk RET mutations compared with more “classic” descriptions of MEN2, although age at onset is highly variable even within the same codon mutation. Even without long-term follow-up, our data do support observations from other studies that many patients have a more indolent disease course. Only 12% of these screened patients had clear lymph node metastases and only 3 (<1%) had distant metastatic disease at presentation. Those with metastases lived 10 or more years with their disease. Additionally, we excluded 40 cases who were obligate carriers because they were not evaluated for MTC; several of these individuals had died at fairly elderly ages of other causes or were alive and apparently healthy, many of them in mid-adulthood. Furthermore, with routine sequencing of codons 10, 11, and 13–16, it has become increasingly common, and sometimes surprising, to encounter lower risk RET mutations in patients presenting with apparently sporadic MTC. In a large series of 481 patients with apparently sporadic MTC, 7.3% were unexpectedly found to carry a germline RET mutation and the distribution of mutated codons differed between patients with a familial versus apparently sporadic presentation, with ATA-A and ATA-B mutations more commonly found in the latter (68). The finding of lower risk RET mutations in apparently sporadic MTC cases highly suggests that certain mutations may be associated with indolent MTC, which could explain an apparent lack of family history of the disease.
Other limitations of this study are its retrospective design and the large proportion of patients ascertained from literature reports, with the potential for incomplete data and varying definitions from different institutions. However, obtaining a prospectively collected adequate sample size from a single institution is unrealistic given the rarity of the disease. We contend that while limitations of this study must be considered in analysis of the data, the information is still valuable. Additionally, the lack of long-term follow-up data in this population precludes the ability of this study to recommend an ideal age for early thyroidectomy. There is also a possibility for publication bias in this study (i.e., extremely benign or aggressive cases preferentially being published), given that most (89%) of our population was comprised of patients from case reports. It is also possible that some patients may have been included more than once in this study despite the care taken to compare studies from the same institution, as patients may have been seen at more than one institution.
We also acknowledge that this study does not establish the true biological onset of MTC in lower risk RET mutation carriers and the extent of MTC varied in our study. Because of this and lack of long-term outcome data, we cannot recommend the ideal approach to management of risk of MTC in presymptomatic ATA-A and ATA-B mutation carriers. Nonetheless, the data derived from this study provide a practical observation of how often MTC is found by age in a “real world” setting, and may be quite valuable in discussions regarding timing of thyroidectomy with patients who have no clinical evidence of MTC. For example, while the likelihood of MTC by age 70 in a codon 611 mutation carrier is estimated to be 89%, the likelihood by age 20 is only 10%. These risks might be interpreted completely differently by parents in the situation of trying to make decisions about the timing of thyroidectomy for their child who has no clinical evidence of MTC. One patient may find a risk of 10% unacceptable and would rather opt for early thyroidectomy, while another may see it as an acceptable risk in the larger context of the risks of surgical complications, consequences of life-long thyroid hormone replacement, and the effectiveness of calcitonin at detecting clinically relevant MTC, preferring to delay surgery until the risk of MTC is higher. Additionally, patients may draw, often incorrectly, from their own, their family members', or members of Internet-based support groups' experience with MTC as representative of all patients with MEN2. Patients with a severely affected relative may perceive their risk to be higher than it actually is, whereas patients whose relatives have had an excellent outcome may underestimate their risk. Patients with a codon 804 mutation may make incorrect assumptions about cancer progression and prognosis from other acquaintances with a codon 620 mutation. The data in this study may help to clarify risk misconception. Risk presentation and engaging patients in discussion about preferred timing of thyroidectomy may result in improved patient satisfaction with their healthcare decisions.
To summarize, this study supports the idea of an individualized, “codon-based” approach to the management of MTC risk in ATA-A and ATA-B mutation carriers that is coupled with clinical data such as calcitonin levels. It provides further evidence that many patients with a lower risk ATA RET mutation have a relatively indolent disease course and provides a better understanding of the age-related progression of MTC. Codons 618 and 620 may need to be classified differently from other ATA-B mutations, and the appropriateness of thyroidectomy in carriers of a codon 791 mutation who have no clinical evidence of MTC should be questioned. Additional study is needed to determine the ability of preoperative calcitonin to predict long-term outcomes in this setting.
Footnotes
Acknowledgment
This research was supported in part by an American Cancer Society Mentored Research Scholar Grant for MEN2 (121138MRSGM1112901) awarded to E.G.
Author Disclosure Statement
None of the authors have a personal or financial conflict of interest.