
Abstract

Resistance to thyroid hormone (RTH) is a syndrome of reduced responsiveness of target tissues to thyroid hormone (TH). It is characterized by elevated TH levels (free thyroxine [fT4] and commonly free triiodothyronine [fT3]), in association with a high or normal thyrotropin (TSH) level, in the absence of intercurrent illness, drugs, or alterations of serum TH transporter proteins. In the majority of cases, the molecular basis of RTH is a mutation in the nuclear TH receptor beta (THRB) gene (1). The estimated prevalence of RTH is 1 in 40,000 (1).
The clinical and biochemical manifestations of RTH are the result of impaired gene regulation by the mutant thyroid hormone receptor (TR) β, as well as interference of the mutant TRβ with the function of the wild type (WT) TRβ. Since the first description of the syndrome in 1967, more than 170 mutations of the THRB gene have been identified (1) but far fewer have been published. This discrepancy is the result of the requirement for labor-intensive functional testing (mutant receptor: affinity to T3, transactivation properties, interaction with transcriptional co-activators, etc.).
We identified two unrelated patients with clinical and biochemical features of RTH who were found to harbor an identical THRB gene mutation (S350L). Since this mutation had not been identified in the literature, we performed in silico modeling to predict the effect of this mutation on the TRβ receptor.
Modeling of the S350L Mutant TRβ
Computer modeling of the expected effect on the structure of TRβ S350L was performed with SwissPDB viewer. The program allows three-dimensional analysis of structural alignments and comparison of active sites or other relevant parts of proteins. In addition, it permits amino acid mutations to be evaluated with simple energy calculations to allow adjustments of the torsional angles of the substituted side chains. Steric collisions, H bonds, angles, and distances are calculated. PDB ID 1XZX was used as a model in the program (2). X-ray crystallography shows the position and environment of S350 in the human TRβ. For example, in PDB ID: 1XZX, S350 is 15 Å from the hormone, and 35 Å from the AF2 coregulator binding site (Fig. 1A). It is found at the start of alpha helix 8 of the receptor, with its side chain pointing inward, away from solvent. The side chain of this amino acid residue forms a hydrogen bond with the main chain carbonyl oxygen of R338, presumably stabilizing the local structure.
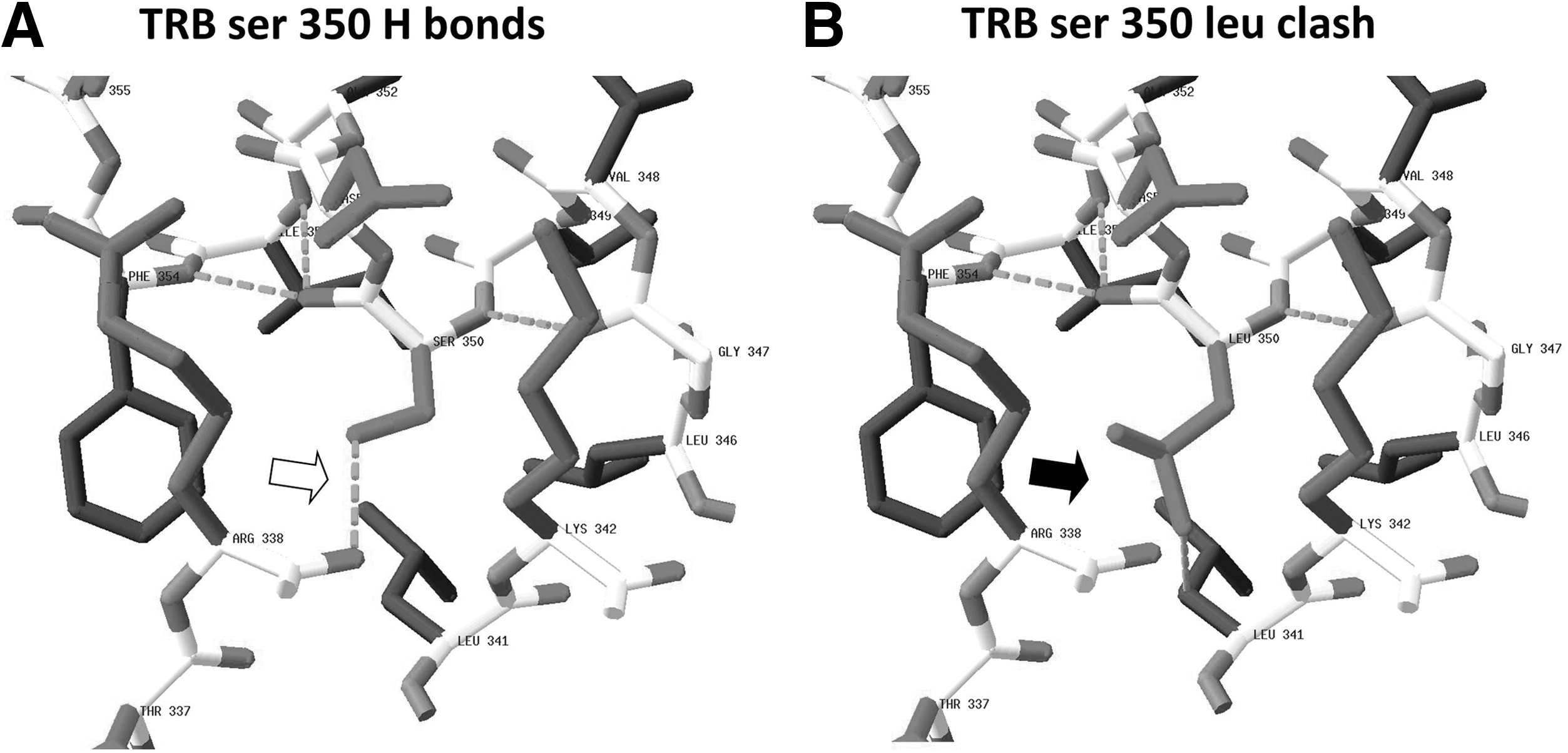
In silico analysis of the S350L substitution in mutant thyroid hormone receptor (TR) β.
Substitution of a leucine side chain for the serine side chain introduces hydrophobic atoms to this mixed polar plus nonpolar environment. By testing all low energy conformers of the leucine side chain, with SwissPDB viewer, we learn that the least compromised conformer of the side chain positions the C delta methyl closer than 2.5 Å to both the carbonyl oxygen of R338 and the C beta methylene of L341. The model predicts that due to steric clashes, the main chain of amino acid 350 and its immediate neighbors would have to move in the order of 1 Å to accommodate leucine at position 350. The stabilizing hydrogen bond between residues 350 and 338 cannot be formed (Fig. 1B).
It is known that mutations in exons 7 to 10 of THRB result in functional impairment of the TRβ protein. In our patients, the location of the mutation in exon 9 of the THRB gene along with the clinical diagnosis of RTH supports causation but does not prove it. In silico modeling predicts that the mutant protein is likely to be less stable than the WT. If this is correct, the on/off rates of coregulators and TH could potentially be affected and some physiological functions of the receptor impaired. Furthermore, via targeted mutagenesis, R338 has been shown to be a member of a cluster of charged, interacting residues whose spatial arrangement is important for stable T3 binding and for optimal homodimer formation on DNA(3). One can therefore speculate as to the significance of the absence of the stabilizing hydrogen bond between residues 350 and R338 due to the S350L mutation, as predicted by SwissPDB viewer.
Despite the absence of functional proof of a causal relationship of this newly identified S350L mutation with RTH, the finding of this mutation in three unrelated patients (4) with RTH is unlikely to represent a chance association.
Footnotes
Author Disclosure Statement
All authors have nothing to declare. No competing financial interests exist.