
Abstract
Background:
Novel small molecular ligands (SMLs) to the thyrotropin receptor (TSHR) have potential as improved molecular probes and as therapeutic agents for the treatment of thyroid dysfunction and thyroid cancer.
Methods:
To identify novel SMLs to the TSHR, we developed a transcription-based luciferase-cAMP high-throughput screening system and we screened 48,224 compounds from a 100K library in duplicate.
Results:
We obtained 62 hits using the cut-off criteria of the mean±three standard deviations above the baseline. Twenty molecules with the greatest activity were rescreened against the parent CHO-luciferase cell for nonspecific activation, and we selected two molecules (MS437 and MS438) with the highest potency for further study. These lead molecules demonstrated no detectible cross-reactivity with homologous receptors when tested against luteinizing hormone (LH)/human chorionic gonadotropin receptor and follicle stimulating hormone receptor–expressing cells. Molecule MS437 had a TSHR-stimulating potency with an EC50 of 13×10−8 M, and molecule MS438 had an EC50 of 5.3×10−8 M. The ability of these small molecule agonists to bind to the transmembrane domain of the receptor and initiate signal transduction was suggested by their activation of a chimeric receptor consisting of an LHR ectodomain and a TSHR transmembrane. Molecular modeling demonstrated that these molecules bound to residues S505 and E506 for MS438 and T501 for MS437 in the intrahelical region of transmembrane helix 3. We also examined the G protein activating ability of these molecules using CHO cells co-expressing TSHRs transfected with luciferase reporter vectors in order to measure Gsα, Gβγ, Gαq, and Gα12 activation quantitatively. The MS437 and MS438 molecules showed potent activation of Gsα, Gαq, and Gα12 similar to TSH, but neither the small molecule agonists nor TSH showed activation of the Gβγ pathway. The small molecules MS437 and MS438 also showed upregulation of thyroglobulin (Tg), sodium iodine symporter (NIS), and TSHR gene expression.
Conclusions:
Pharmacokinetic analysis of MS437 and MS438 indicated their pharmacotherapeutic potential, and their intraperitoneal administration to normal female mice resulted in significantly increased serum thyroxine levels, which could be maintained by repeated treatments. These molecules can therefore serve as lead molecules for further development of powerful TSH agonists.
Introduction
S
Thyrotropin (TSH) is a heterodimeric glycoprotein hormone secreted from the anterior pituitary and mediates its action through the TSHR, which is a member of the class A GPCR family. The holoreceptor consists of 764 amino acids divided into three regions. The first is a large, highly glycosylated ectodomain of which the initial 260 amino acids incorporate 10 leucine-rich repeats and which has been crystallized bound to a stimulating TSHR antibody (3). The second part of the ectodomain is a region of 130 amino acids and is known as the signal-specific domain (SSD) or “hinge region” (4 –6) incorporating two additional leucine-rich repeats and a unique 50 amino acid insert. A partial homology model of this enigmatic hinge connecting the ectodomain to the third part of the receptor—the transmembrane domain (TMD)—has been derived from the recently crystallized follicle stimulating hormone (FSH) receptor hinge region (7). The TMD consists of 349 amino acids typical of the GPCR class A family incorporating seven transmembrane helices (TMH) joined by extracellular (ECL) and intracellular (ICL) loops (8).
The TSHR is primarily expressed in the thyroid to carry out its physiologic role in thyroid cell growth and hormone synthesis/secretion, but it also happens to be a primary autoantigen in autoimmune thyroid disease, especially Graves' disease (8 –10). In addition to its primary site on the thyroid cell, the TSHR is expressed in a variety of extrathyroidal tissues where it is known to modulate target cell function (9). For example, the roles of the TSHR in Graves' orbitopathy, adipogenesis, and bone metabolism have been extensively studied (11 –13). Therefore, modulating the function of the receptor either orthosterically using monoclonal antibodies (14) or allosterically, in either a positive or negative fashion, using SMLs (15 –17) has therapeutic potential. Investigations by Neumann et al. using chemical modification of reported SML agonists of the luteinizing hormone/chorionic gonadotropin receptor (LH/hCGR) (18) have resulted in small molecule agonists and antagonists to the TSHR in the nanomolar range (15,16), while other reports of potent small molecular antagonists to the TSHR lacked receptor specificity (19). Thus, there remains a need to develop higher potency small molecule agonists and antagonists to the TSHR to develop into therapeutic drugs.
Molecular modeling studies have provided considerable insight into the mode of action of these small molecule agonists (20). SMLs by virtue of their small size can permeate the cell and dock with polar and nonpolar residues in the TMD region and exert their effects in an allosteric manner (21). In the case of the TSHR, the different transmembrane helices and intracellular and extracellular loops connecting these helices have been studied extensively for activating and inactivating mutations (22), which are the “hot-spots” in the receptor, having been found in human pathological conditions (21). Based on such modeling and mutational analysis of TSHR “hot-spots,” there appear to be two clusters of residues spanning the TM helices that are preferred mutation sites, as well as potential sites for docking of selected small molecules (20,23). Perturbations in and around the vicinity of these “hot-spots” are thought to lead to conformational changes in the intrahelical regions, which can either distort the basal constrained structure of these helices (24) or/and disrupt a polar interaction between a partially conserved motif on the end of TM3 and another conserved polar reside at the base of TM6, leading to disruption or destabilization of the “ionic lock” as seen in most GPCRs, including the TSHR (25 –27).
In this report, we describe the identification and characterization of two new potent and novel small molecule agonists to the TSHR selected using a luciferase based high-throughput screening (HTS) assay. The ability of these lead molecules to activate different G proteins has been studied in addition to their pharmokinetic (PK) characteristics and their ability to cause the release of thyroxine (T4) in treated animals.
Materials and Methods
Materials
Bovine TSH (1 IU/mL), human FSH (70 IU/mL), human chorionic gonadotropin (hCG) (10 IU/vial), and forskolin were purchased from Sigma-Aldrich (St. Louis, MO). The Bright-Glo™ luciferase substrate (Cat # E2610) was purchased from Promega Corp. (Madison, WI). The cell culture medium Dulbecco's modified Eagle's medium (DMEM) and Ham's F12 were purchased from Mediatech, Inc. (Manassas, VA). Fetal bovine serum (FBS) and fetal calf serum were purchased from Atlanta Biologicals (Flowery Branch, GA). Additional amounts of the lead compounds that we have identified in our screen were purchased from ChemBridge Corp. (San Diego, CA). Mouse primary Sertoli cells were obtained from ATCC (Manassas, VA). The L1 library, a collection of 100,000 molecules from Chembridge that satisfy most of the Lipinski's Rule of Five, was obtained from the Integrated Screening Core at the Icahn School of Medicine at Mount Sinai, New York.
Stable cell lines used
CHO-HA-TSHR luciferase cells
For HTS, we used cells generated by transfecting the pGL4.29 [luc2P/CRE/Hygro] construct into a highly selected stable line of CHO cells expressing the human TSHR with a HA (hemagglutinin) tag at the N-terminus (CHO-HA-TSHR cells) that has been previously described (28) and selected as a stable line with hygromycin.
TSHR/LHR chimeric luciferase cells
We used a construct pSV2-neo-ECD-TSH-LHR-11 (kindly provided by Dr. Basil Rapoport, Cedars-Sinai Research Institute and University of California, Los Angeles, CA), where a 367 amino acid insert containing the homologous regions of the rat LH/hCGR sequence replaced the TSHR ectodomain (29), which was then co-transfected with the pGL4.29 [luc2P/CRE/Hygro] construct in CHO cells and further selected for double transfectants.
CHO luciferase cells
These cells were generated by transfecting the pGL4.29 [luc2P/CRE/Hygro] construct into the CHO PSVL cells (JPO2) and selecting with hygromycin for stable transformants. The best stable clone was selected based on different concentrations of forskolin and unresponsiveness to TSH.
Each of these stable cell lines were cultured in Ham's F12 medium with 10% FBS, 100 IU/mL of penicillin, 100 μg/mL of streptomycin, and 50 μg/mL of hygromycin.
Primary Sertoli cells (line TM4)
These FSHR-expressing cells were obtained from ATCC (CRL-1715) and cultured in DMEM: F12 medium (cat # 30-2006) with 2.5% FBS and 5% horse serum (ATCC; cat #30-2040).
LHR-expressing cells
The specificity against the LH/hCGR was tested using a stable line of rat hCGR in HEK 293 cells that we obtained from Dr. K.M.J. Menon (University of Michigan, Ann Arbor, MI).
Stable lines of luciferase constructs for G protein activation
For signaling studies of various G proteins, we used cells generated as previously described (30). We used double-transfected stable lines of CHO-HA-TSHR with pGL4.34 [luc2P/SRF-RE/Hygro] to detect Gα12, pGL4.33[luc2P/SRE/Hygro] to detect Gβγ, and pGL4.30 [luc2P/NFAT-RE/Hygro] to detect Gαq. These double-transfected stable lines were also maintained in complete Ham’ F12 medium with appropriate concentrations of hygromycin.
HTS luciferase assay
To develop the screening assay, a high expressing stable line of CHO-HA-TSHR cells carrying an amino terminus HA tagged TSHR was selected. This stable CHO line was transfected with the construct pGL4.29 [CRE/minP/luc2P] carrying a minimal promoter driving a CREB response element (CRE) tagged to a modified form of the luciferase reporter gene luc2P (Promega Corp.). Luc2P is a modified firefly luciferase sequence with humanized codon optimization that is designed for high expression and reduced anomalous transcription. In addition, the luc2P gene contains hPEST, a protein destabilization sequence, which further reduces background transcribed protein (31). Activation of the TSHR by TSH or an agonist results in Gsα-adenylate cyclase coupling and an increase in intracellular cAMP, which results in the activation of CREB at its binding to the CRE element and subsequently in the transcription of the luciferase gene and accumulation of the luciferase enzyme within the activated cells. Luciferase activity in these cells was detected after lysing the cells using the commercial substrate Bright-Glo™. For HTS, we seeded 15,000 cells of transfected CHO-HA-TSHR cells per well in a 384 opaque white-bottom poxi-plate (ProxiPlate cat # 6008230; PerkinElmer, Branford, CT) using a Multidrop Combi dispenser (Thermo Fisher, Waltham, MA) in 10 μL of Ham's F12 complete medium, and incubated overnight at 37°C in a CO2 incubator with a relative humidity of >85%. Small molecule libraries were added from 384-well stock plates containing 10 mM solutions in DMSO (Chembridge Corp.). Small molecule addition was accomplished using a 384-tip pin tool (V&P Scientific, San Diego, CA) transferring 17 nL per pin (based on fluorimetric calibration), resulting in a final concentration of 17 μM per well. Plate validation and normalization controls, including negative control (medium only) with 0.1% DMSO and a positive agonist molecule previously described (15), were added to blank wells located in the first two and last two columns of each plate. Following compound addition, plates were incubated for 4 h at 37°C. After 4 h, the cells were lysed by adding 6 μL of Bright-Glo™ reagent, and incubated for 2 min before measuring luciferase activity using an EnVision Multilabel Plate Reader (PerkinElmer). Throughout the screen of 137 plates, the signal-to-background ratio was linear, the mean CV was 12%, and the Z′ factor was in the range 0.7–0.8 based on the positive control used in the plate, which exceeds the commonly accepted threshold (0.5) for validation of high-throughput assays (32). Dose–response relationships of the lead molecules were determined using a Tecan HP dispenser by following a similar protocol. Data points of the dose–response curves were fitted using Prism 5.0.
Docking of lead molecules on the TSHR transmembrane
Docking of the lead molecules was performed on a homology model of the TSHR-TMD based on rhodopsin (PDB:1F88). This template was chosen because of the low RMSD values between the backbone of the TM helices of the TSHR model and that of the rhodopsin x-ray crystal structure (33), and fits the experimental parameters previously described (34). The initial homology model of rhodopsin was obtained from the Uniprot server (
Molecular dynamics simulation of lead molecules
To assess the quality of the binding pocket further, we performed a 10 ns full-scale molecular dynamics simulation of bound poses of the lead molecules using the AMBER software suite v12 that uses force field 12SB (41). The initial docked conformation as analyzed for binding sites was obtained by DOCKRES (40). All the force field parameters of the two lead compounds interacting via only nonbonded parameters with the receptor were obtained by Antechamber tools using Gaussian calculations. The topology and initial coordinates of the lead molecules forming complexes with TSH-TMD were created with the AMBER LEAP module (41).
Molecular dynamics on both complexes was carried out under periodic boundary conditions (PBC) with 10 Å nonbonded cutoff lengths. A total of 1000 minimization steps composed of 500 conjugate gradient and 500 steepest descent were performed followed by the heating cycles. The first heating cycle of 5000 steps was followed by a heating cycle of 50,000 steps that equilibrated the system temperature to a simulation temperature of 300 K. The final MD simulation of the complexes at constant pressure and temperature were carried out for 10 ns with a 2 fs integration step size. The trajectories were analyzed and visualized by Simulaid and VMD (39,42). The molecular simulation indicates general stability and affinity of both the lead molecules to the binding pockets as predicted from the docking results.
Gene expression studies by quantitative reverse transcription polymerase chain reaction
Total RNA was isolated from FRTL5 cells treated with 1 μM of MS437 for 4 h and untreated control cells using TRIzol reagent (Invitrogen, Life Technologies, Carlsbad, CA) in accordance with the manufacturer's instructions. The RNA purity was evaluated by the ratio of absorbance at 260:280 nm (>1.9). After digestion of genomic DNA by treatment with TURBO DNA-free™ DNase I (Ambion, Austin, TX), total RNA (1 μg) was reverse-transcribed into cDNA with random hexamers using Advantage RT-for-PCR kit (Clontech, Mountain View, CA). The quantitative reverse transcription polymerase chain reactions (qRT-PCRs) were performed using the StepOnePlus Real-time PCR system (Applied Biosystems, Foster City, CA). The reactions were established with 10 μL of SYBR Green master mix (Applied Biosystems), 0.4 μL (2 μM) of sense/antisense gene-specific primers, 2 μL of cDNA, and DEPC-treated water to a final volume of 20 μL. The PCR reaction mix was denatured at 95°C for 60 s before the first PCR cycle. The thermal cycle profile was as follows: denaturizing for 30 s at 95°C, annealing for 30 s at 57–60°C (dependent on primers), and extension for 60 s at 72°C. A total of 40 PCR cycles were used. For each target gene, the relative gene expression was normalized to that of the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) housekeeping gene by use of the Applied Biosystem StepOnePlus Real-time PCR systems software. Data presented (mean) are from two independent experiments in which all sample sets were analyzed in triplicate.
Mouse thyroid function testing
Female C57BL/6J mice (Jackson Laboratory) six to eight weeks of age and with a mean body weight of 20–25 g were maintained on a standard diet and received intraperitoneal (i.p.) injections (100 μg/mouse of MS437/MS438) for three consecutive days in a fluid volume of 60–90 μL containing a final concentration of ∼25% DMSO. The control animals received diluted vehicle (DMSO) or bovine TSH 30 μg/mouse by the same route. Thyroid hormone (T4) levels were estimated in serum from blood collected by submandibular bleeding prior to treatment (prebleed) and 72 h post-treatment (postbleed). Total T4 was measured by a neonatal T4 RIA kit (Coat-A-Count, Siemens Medical Solutions Diagnostics, Berkeley, CA) according to the manufacturer's protocol. All experiments involving animals were carried out according to the Institutional Animal Care Committee Guidelines.
G protein signaling studies
As outlined before, we developed stable CHO-HA-TSHR cell lines expressing various reporter vectors (CRE-, NFAT-RE, SRE-, and SRF-RE-) for studying the activation of various G proteins. These stable lines were characterized and optimized for responses using positive (TSH, ionomycin, PMA) and negative controls. Prior to measurement of signaling, 20,000 cells were seeded in square-bottom white plates (Nunc cat # 164610) in 20 μL of Ham's F12 complete medium and incubated overnight at 37°C. The complete medium was then replaced with serum free medium for 2 h and then treated with 10 μM of each compound and the appropriate controls for 4 h. At the end of the incubation period, the cells were lysed using 10 μL of Bright-Glo™ reagent and incubated for 2 min at RT, and the plates were read using a BMG Pherastar microplate reader.
PK studies
PK studies were carried out at Sai Life Sciences Ltd. (Pune, India). Briefly, 18 Balb/c mice were used for testing each molecule. The animals were weighed before dose administration and divided into two groups. Group 1 was bolus-dosed intravenously (i.v.), and group II was bolus-dosed via i.p. injection with careful formation of a solution at a dose of 20 mg/kg body weight. Blood samples were collected at predose, and at 2, 4, 6, 12, and 24 h (i.v. and i.p.). Blood was collected from a set of three mice under light isoflurane anesthesia from the retroorbital plexus at each time point in tubes containing K2EDTA as anticoagulant. Plasma samples were processed for analysis by protein precipitation using acetonitrile. Glipizide was used as internal standard and analyzed by the LC-MS/MS method. PK parameters were calculated using the noncompartmental analysis tool of Phoenix Win Nonlin Enterprise software (v6.3).
Statistical analyses
All curve fitting and EC50 calculations were performed using GraphPad Prism v5.02, and statistical differences for p-values were calculated using one-way analysis of variance (ANOVA) using Prism.
Results
Identification of positive allosteric modulators of the TSHR
To identify allosteric modulators of the TSHR, we screened 48,224 small molecules from a 100K library using the one-step transcriptional-based bioassay. All compounds were screened at a single concentration of 17 μM in duplicate plates and selected if a significant response was obtained in both plates and with the selection criteria being more than three standard deviations (SD) above the baseline activity. The screen of 48,224 molecules resulted in a total of 63 positives, giving a hit ratio of 0.13%. False positives are commonly found in any cell-based signaling assays. Therefore, to identify true agonist compounds, we subjected the most positive 22 of the 63 hits to a second confirmatory testing using TSHR-expressing CHO cells and also CHO cells containing an empty vector (parent cells). Based on this second round of testing, we selected two compounds—MS437 and MS438—as our lead molecules, which showed >10-fold responses above the baseline and no activity on parent CHO luciferase cells. Figure 1A and B shows the structures of these lead molecules, and Figure 1C and D shows the dose–response curves for TSH and lead molecules. Molecule MS437 had an EC50 of 13×10−8 M, and molecule MS438 had an EC50 of 5.3×10−8 M.
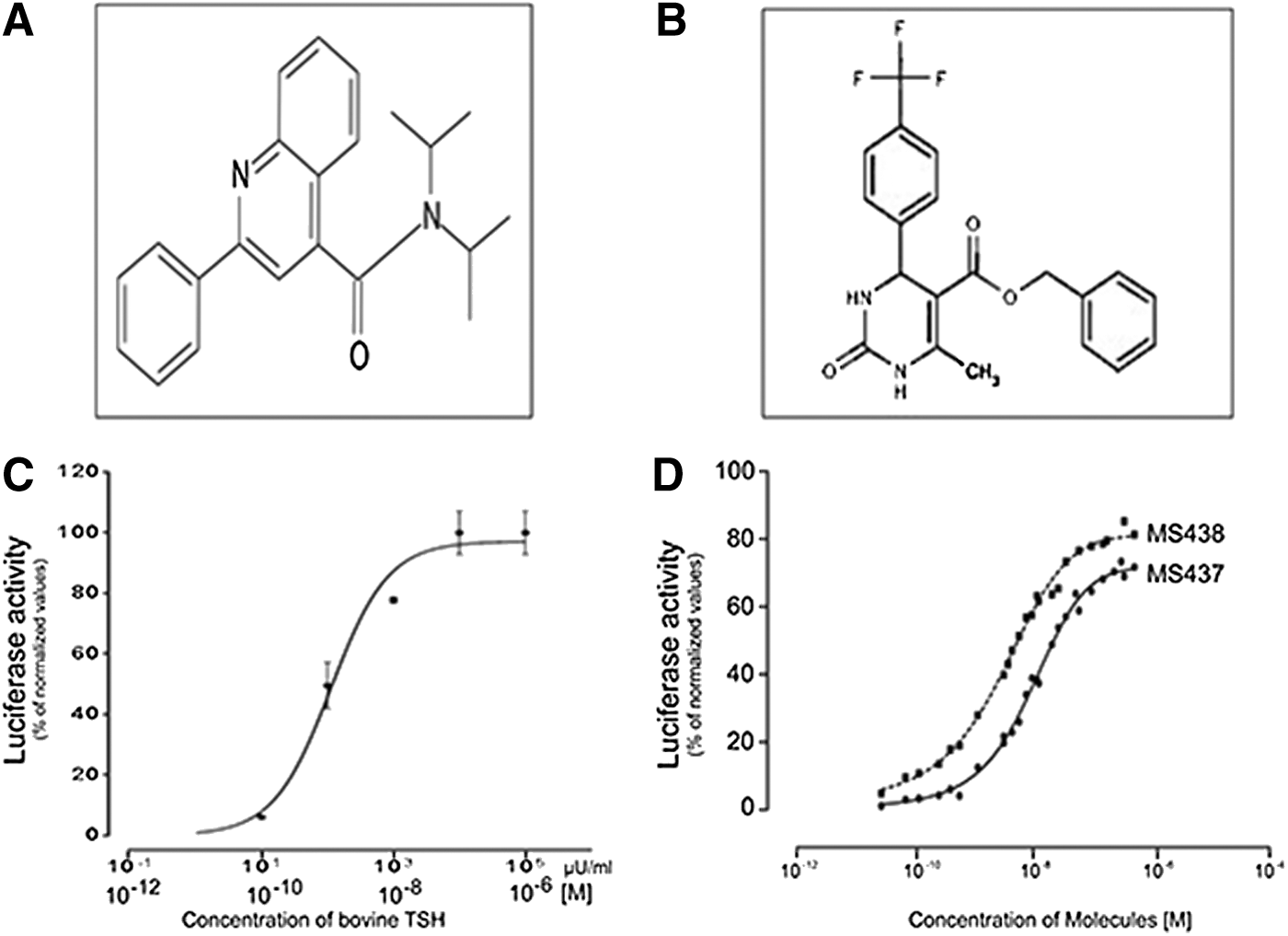
Structure and potency of selected lead molecules. (
Specificity of lead molecules
To analyze the specificity of these molecules, the compounds were tested against cells that expressed the LHR or the FSHR. For the LHR cells, we used HEK 293 cells transfected with the rat LH/hCGR, and for FSHR cells, we used primary murine Sertoli cells (line TM4), which express the FSHR and respond to human FSH in a dose-dependent manner. Intracellular cAMP was measured after stimulation with 0.1, 1, and 10 μM of the lead molecules and corresponding positive and negative controls. The two lead molecules failed to show any activity against the LHR or the FSHR-expressing cells, even at the highest concentration used (10 μM); in contrast, they responded to treatment with their respective ligands hCG (1000 μIU/mL) and human FSH (700 μIU/mL) incorporated as positive controls (Fig. 2).
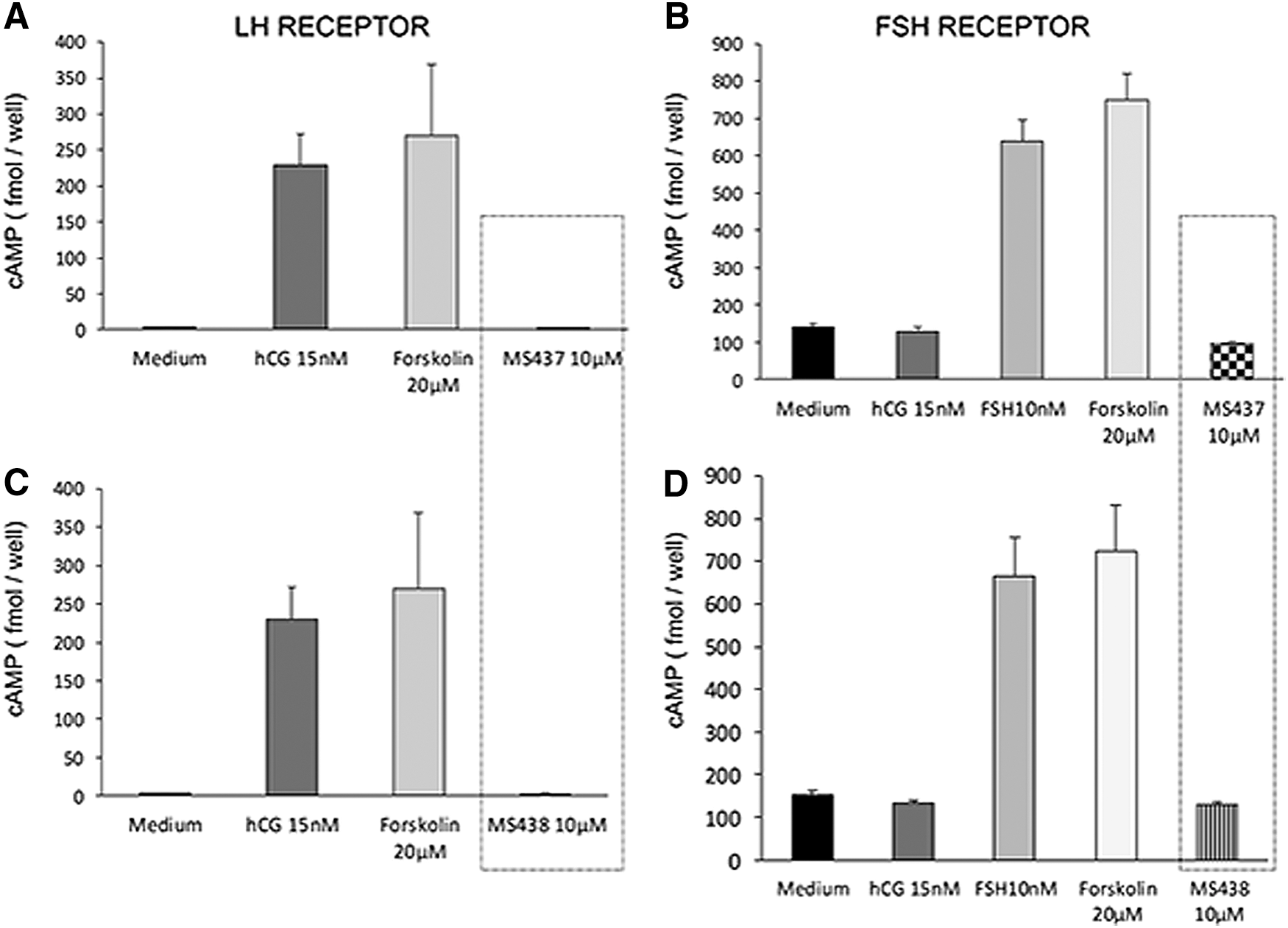
Specificity of the molecules. (
Defining the binding sites
Small molecules are known to often activate GPCRs in an allosteric manner by binding to the TMD of the receptor (43). To examine if molecules MS437 and MS438 bind to the TMD of the TSHR, we used a chimeric construct as schematically represented in Figure 3A. In this construct, the TSHR ectodomain is replaced with the LHR ectodomain, but it retains the complete TSHR TMD (29). Stable cells co-transfected with this chimeric receptor and a luciferase construct responded to hCG (1000 μIU/mL) but not to recombinant human TSH (3000 μIU/mL) indicating the specificity of the ligand binding ectodomain. On exposure to 10 μM of MS437 and MS438, the cells showed equivalent or greater responses compared to hCG and forskolin (Fig. 3B top and bottom panels), indicating that the molecules bound to the serpentine portion of the TSHR.
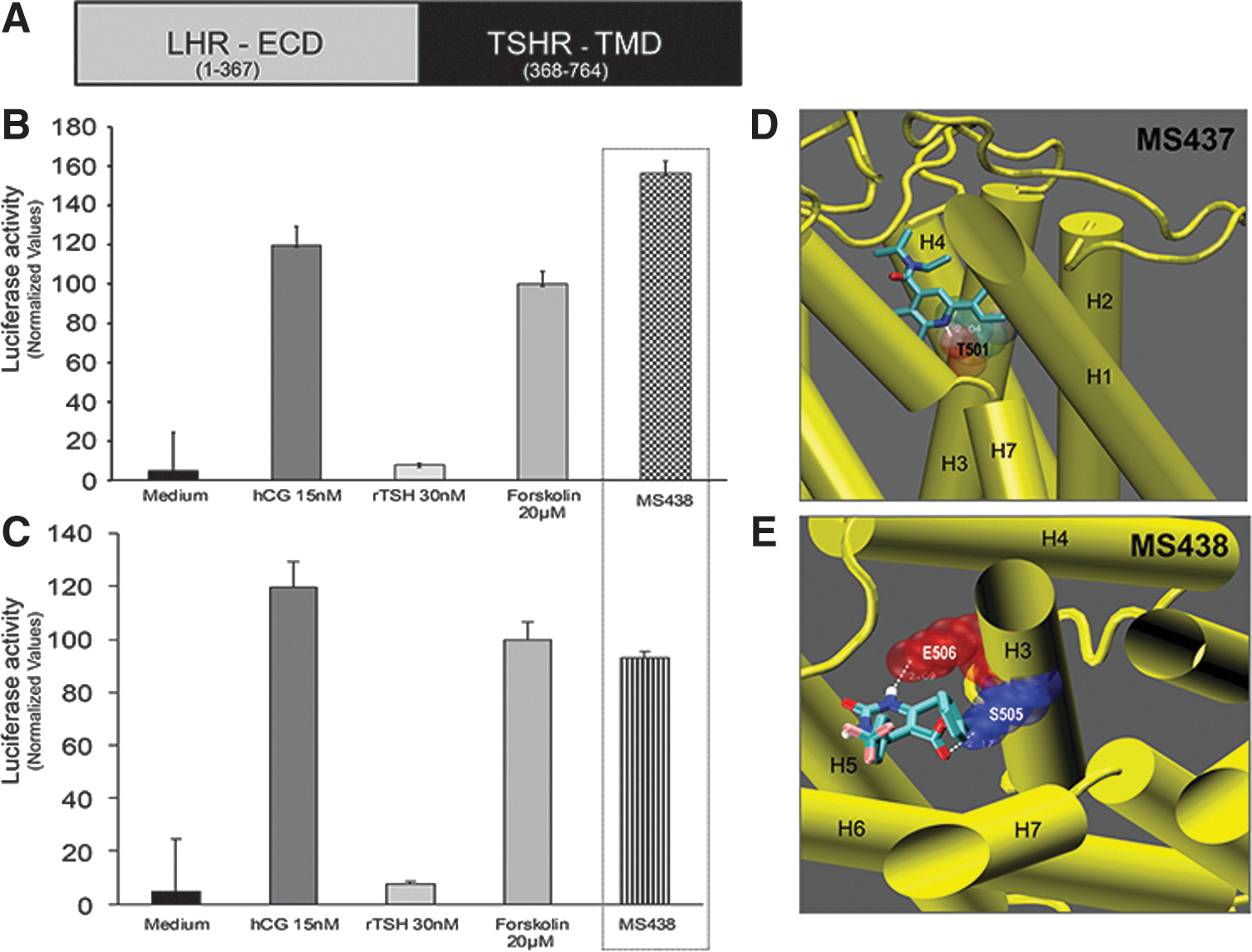
Allosteric activation of the TSHR. (
To identify the binding pocket(s) of the lead molecules, they were docked to the TM region using a structure of the TSHR TM region developed in our laboratory by homology modeling based on the rhodopsin crystal structure. By docking these molecules using Autodock, we were able to show their putative interaction with residues in the intrahelical region of TM3. As per this docking, molecule MS437 interacts with threonine 501 (T501) of TM3 (Fig. 3D), and molecule MS438 interacts with residues serine 505 (S505) and glutamic acid 506 (E506) respectively (Fig. 3E). A preliminary molecular dynamics simulation of just the TMD and the docked ligand showed no tendency of the ligands to leave their site.
Classical and nonclassical G protein signaling studies
The TSHR has been reported to activate members of all four G protein families (Gsα, Gq/11, Gβγ, and G12/13) (44). We studied the signaling potential of our two lead small molecules using a quantitative technique via the tagged response elements for CRE, SRE, SF-SRE, and NFAT (30). Using these constructs, the major pathway that was activated by MS437 and MS438 was the classical Gsα pathway where we saw a robust and significant response similar to TSH as indicated in Figure 4A. Examining the other G proteins for nonclassical pathway responses, we observed that the MS437 and MS438 molecules were able to activate Gq/11 by increasing NFAT and Gα12 by stimulating a SRE luciferase reporter (Fig. 4B and C). Neither MS437 nor MS438 showed any significant activation of RhoA kinase via SRF luciferase, indicating that the small molecules did not engage Gβγ (Fig. 4D). The activation of Gsα and Gq by the small molecules, similar to TSH, strongly suggests that these molecules could initiate iodine organification and thyroid hormone secretion and promote thyroid growth by their ability to engage in Gq activation in the same manner as TSH itself (45 –48).
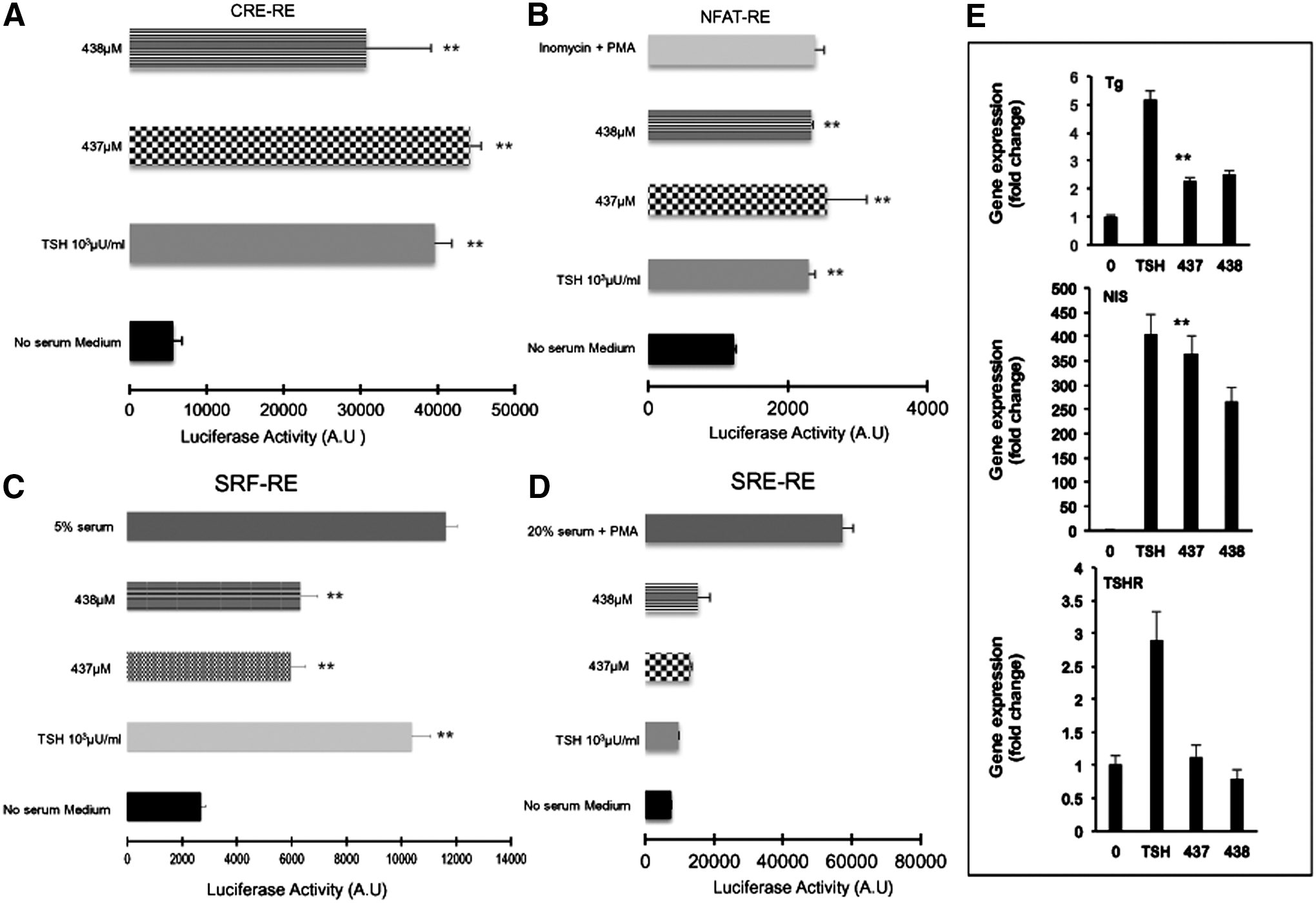
TSHR signaling via different G proteins. In order to study the engagement of different G proteins with the TSHR, we generated stable CHO cells containing various luciferase reporter constructs with distinct response elements as detailed in Materials and Methods. (
Thyroid-specific gene expression in thyrocytes
In order to confirm the activity of these molecules on more physiologically relevant cells, we examined thyroid-specific gene expression using rat thyrocytes (FRTL5). We tested the effects of these two molecules on expression of mRNA of the thyroglobulin (TG), sodium-iodide symporter (NIS), thyroid peroxidase (TPO), and the TSHR genes. Prior to exposure, the cells were deprived of TSH for 48 h and starved in serum-free medium for another 2 h. A single-dose treatment of 1 μM of MS437 and MS438 for 4 h showed a two- to eightfold increase in thyroid-specific gene expression (Tg, NIS, and TSHR) when measured by qPCR. These data show that the SMLs had the ability to exert their effects on thyrocytes that express relatively low levels of the TSHR compared to the previously transfected cell lines (Fig. 4E).
In vivo potency of the lead molecules
In order to show that these lead molecules are potential candidates to develop clinically relevant drugs, we tested their ability to cause release of thyroid hormones in vivo. T4 levels were measured in male C57BL/6J mice after three i.p. bolus injections of 100 μg per mouse per day (total of 0.3 mg per mouse) of MS437 and MS438 dissolved in<50% DMSO. In this protocol of prolonged treatment, we saw a sustained increase in serum T4 levels (Fig. 5A). This in vivo T4 release study clearly indicates the potency of our lead molecules as agonists to the TSHR.
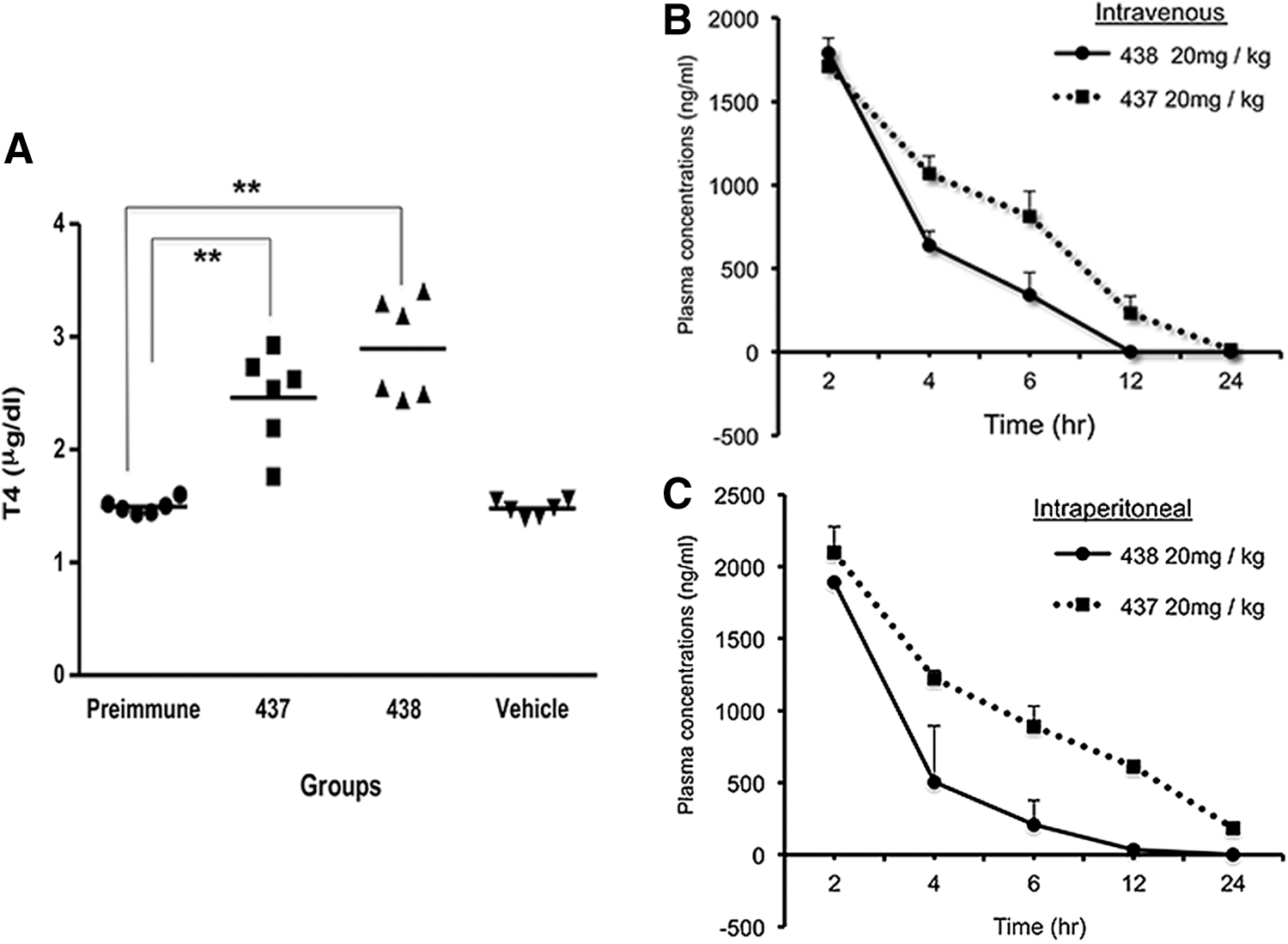
Small molecule-induced T4 secretion and pharmacokinetics. (
PK studies
The in vivo potency studies allowed us to pursue standard PK analyses of the two lead molecules. As described, a single bolus injection of each compound at a dose of 20 mg/kg was given to Balb/c mice (weight 20–35 g each, ∼0.4 mg per mouse) by i.v. and i.p. routes, and the murine plasma was analyzed by mass spectrometry at different points after reaching T max (Fig. 5B and C). Although the time course of clearance was similar by both routes of administration, we observed a half-life (T 1/2) of 3 h for MS437 compared to a shorter half-life (T 1/2) of 1 h for MS438 when measuring the half-life of the lead molecules. However, the T 1/2 for both routes of administration remain the same, as T 1/2 is calculated from the terminal elimination points. Both compounds show moderate plasma clearance (MS437=24.13 mL/min/kg and MS438=29.63 mL/min/kg), high volume of distribution (MS437=9.6-fold higher vs. MS438=4.6-fold higher than total body water) indicating extravascular distribution. In particular, compound MS437 fits into a good PK parameter category with high exposure after i.p. administration.
Discussion
HTS approaches play a central role in identifying small molecules that have intrinsic activity (49), and this approach has been used to identify molecules targeting the TSHR. However, the first reported agonists to the TSHR were identified based on chemical modification of thienopyrimidine org41841, which was previously identified as an agonist to the LH/hCGR (18,50). Subsequent successful agonists and antagonists to the TSHR reported by Neumann et al. (51) have been obtained by a combination of HTS and meticulous chemical modifications. The reported agonist compound 2 (52) stimulated thyroid-specific gene expression in primary human thyrocytes and produced thyroid hormone in mice after administration of 0.5 mg of the molecule. We ventured to identify additional small molecule candidates against the TSHR with the intention of developing greater or unique activities. In this report, we describe the identification and characterization of two novel small molecule agonists detected using a new high-throughput assay.
Our high-throughput strategy employed a remarkably sensitive and precise one-step transcriptional-based assay for measuring interaction of TSH or small molecules with the TSHR. In this system, cAMP responses modulated by the binding of TSH or small molecules were detected as luciferase generation in transfected heterologous co-expressing the TSHR and a CRE-luciferase construct. Initial optimization and validation of the assay gave robust Z′ scores (Z′=0.6–0.8), Z′ being a parameter for the measurement of the robustness of a high-throughput assay (32). Using a 384-well format, we screened 48,224 compounds, and found 63 molecules that showed activation of the TSHR. These “hits” were further refined and resulted in the selection of two molecules showing consistent and vigorous dose–response relationships with an EC50 in the nanomolar range similar to the previous report (15) and approximately equipotent to bovine TSH. Furthermore, these molecules had structures that were dissimilar to any TSHR agonists reported so far (15) (Fig. 1). We refer to these two lead agonist molecules as MS437 and MS438.
Having shown no interaction with the parent control cells (CHO luciferase), which express a variety of receptors including adrenergic receptors, our next approach was to test the specificity of the lead molecules by examining their interaction with the LH/hCGR and the FSHR. The two lead molecules, even at 10 μM (10−5 M) concentrations, caused no response with cells expressing the LH/hCGR or cells expressing the FSHR (Fig. 2) as found previously (15). This is in contrast to an earlier TSHR agonist active in nanomolar concentrations but which had clear specificity spillover (19). Although the lead molecules did not interact with the receptors tested, including the many receptors expressed on control CHO cells, the GPCR family of receptors is large, and we cannot exclude any interactions at this time. Thus, our two lead molecules—MS437 and MS438—appear to have selectivity and potency for the TSHR and have efficacy equivalent to TSH in cells expressing the TSHR.
Small molecule agonists and antagonists, unlike TSH, have been shown to mediate their signaling effects by binding to the TMD of the TSHR (20). This allosteric mode of action may result from the inherent property of small molecules, including their small size and hydrophobic nature. However, it has been shown that the TSHR contains clusters of residues with allosteric potential encompassing signaling-sensitive amino acids within the helices of the TMD (21). To determine if our lead molecules were allosteric modulators, as expected, we tested their receptor-stimulating activity on a chimeric receptor in which the major part of its ectodomain consists of the LHR sequence fused to the native human TSHR TMD. The two lead molecules activated luciferase generation with this chimeric receptor expressed in parental CHO cells but were unable to activate the native LH/hCGR as described earlier. Hence, the molecules required the TSHR TMD in order to signal, showing that they bind to the serpentine domain of the receptor. This is in great contrast to TSH, which exerts its effect by binding mainly to epitopes in the LRD of the ectodomain but also has other binding sites in the large extracellular domain and extracellular loops (53,54).
To substantiate our in vitro findings and to map the binding sites of these molecules, we used a new homology model of the TSHR TMD with its loop structures. These docking studies predicted that both molecules bind to the intrahelical region of the transmembrane helix 3 (TMH3) and result in hydrogen bonding with residues T502 for MS437 and E505 and E506 for MS438 of TMH3 (Fig. 3); this contrasts with the binding of an earlier reported agonist to TMH5 (15). Preliminary molecular dynamic simulation studies carried out for 10 ns showed that binding of these molecules to TMH3 was of high affinity, allowing them to be retained in their binding pocket.
The potential of a small molecule as a drug candidate lies primarily in the selective activation or inhibition of the receptor. Although it has been shown that the TSHR is capable of engaging multiple G proteins (55), recent studies from our laboratory using different autoantibodies to the TSHR (56) have shown that engaging different epitopes of the receptor can result in a cascade of differential signaling via classical (cAMP-PKA) and nonclassical (MAPK/Rho) signaling pathways (47,57). Furthermore, such signaling may alter the immunopathology of autoimmune thyroid disease (58). Using a luciferase reporter that was transcriptionally activated by a cascade of downstream messengers induced by different G proteins, we studied Gsα, Gαq, Gβγ, and Gα12 activation by the lead small molecules. Like TSH, the two lead agonists primarily engaged Gsα, Gαq, and Gα12, resulting in endogenous gene expression of the Tg, NIS, and the TSHR genes (Fig. 4A–D).
To be potential drug candidates, the small molecules need to display in vivo activity, and we show their ability to stimulate the thyroid gland and increase secretion of T4 in mice. The PK studies indicate that both lead molecules have good PK parameters with moderate plasma clearance and half-life, and both are capable of enhancing thyroid hormone secretion. Furthermore, in vitro cytotoxic studies showed no cytotoxic effects at the highest working concentrations (data not shown) of these two molecules. Hence, the data show that we have identified two structurally unique lead molecules that are selective allosteric agonists to the TSHR. The molecular scaffolds of these two lead molecules hold promise for chemical optimization and molecular changes to derive even more potent agonists and antagonists to the TSHR.
Footnotes
Acknowledgments
The authors thank Dr. Yaron Tomer and Dr. Mone Zaidi from the Department of Medicine, Icahn School of Medicine at Mount Sinai, for their helpful discussions. We are also thankful to Dr. R. Baliram and Dr. X.Yin for their contributions. The luciferase constructs mentioned in this study were a gift from Dr. Frank Fan, Director of Research, at Promega Corp. We also thank the computational resources and staff expertise provided by the Department of Scientific Computing at Icahn School of Medicine at Mount Sinai, New York.
This work was supported in part by NIH grants DK069713, DK052464, and the VA Merit Award program (to T.F.D.).
Author Disclosure Statement
No competing financial interests exist.