
Abstract
Background:
Critically ill patients typically present with low or low-normal plasma thyroxine, low plasma triiodothyronine (T3), increased plasma reverse T3 (rT3) concentrations, in the absence of a rise in thyrotropin (TSH). This constellation is referred to as nonthyroidal illness syndrome (NTI). Although it is long known that the severity of NTI is associated with risk of poor outcomes of critical illness, the causality in this association has not been well investigated.
Summary:
In this narrative review, the different faces of NTI during critical illness are highlighted. Acute alterations are dominated by changes in thyroid hormone binding, peripheral thyroid hormone uptake, and alterations in the expression and activity of the type-1 and type-3 deiodinases. It was recently shown that at least part of these acute changes are brought about by concomitant macronutrient restriction, and this part appears adaptive and beneficial. However, the face of the NTI in the prolonged phase of critical illness is different, when patients are fully fed but continue to depend on intensive medical care. In that prolonged phase of illness, hypothalamic thyrotropin releasing hormone (TRH) expression is suppressed and explains reduced TSH secretion and whereby reduced thyroidal hormone release. During prolonged critical illness, and in the presence of adequate nutrition, several tissue responses could be interpreted as compensatory to low thyroid hormone availability, such as increased expression of monocarboxylate transporters, upregulation of type-2 deiodinase activity, and increased sensitivity at the receptor level. Infusing hypothalamic releasing factors in these prolonged critically ill patients can reactivate the thyroid axis and induce an anabolic response.
Conclusions:
It is clear that the name “NTI” during critical illness refers to a syndrome with different faces. Tolerating the early “fasting response” to critical illness and its concomitant changes in thyroid hormone parameters appears to be wise and beneficial. This thus applies to the NTI present in the majority of the patients treated in intensive care units. However, the NTI that occurs in prolonged critically ill patients appears different with regard to both its causes and consequences. Future studies should specifically target this selected population of prolonged critically ill patients, and, after excluding iatrogic drug interferences, investigate the effect on outcome of treatment with hypothalamic releasing factors in adequately powered randomized controlled trials.
Introduction
P
NTI occurs in response to fasting in healthy subjects (6,7) and is present in patients suffering from acute severe illnesses (1 –5). NTI in response to fasting in otherwise healthy subjects has shown to be an adaptive and beneficial response in the sense that lowering the availability of the active thyroid hormone T3, via a reduced thyroid hormone production and an immediate thyroid hormone inactivation into rT3, as well as a limitation of the peripheral uptake of thyroid hormones, reduces energy expenditure to limit catabolism (7,8). The pathways by which fasting induces an immediate decrease in active thyroid hormone T3 comprise several changes, among which the immediate suppression of the deiodinase type-1 (D1), the dominant enzyme driving the conversion of T4 to T3, is crucial (6,7). Also an increase in type-3 deiodinase (D3) activity with fasting contributes to the rise in rT3 (6,7). In addition, TSH secretion is suppressed by fasting, brought about by a multifactorial response in which suppressed leptin release could play an important role (6,9). A reduced peripheral uptake of thyroid hormone with prolonged fasting also contributes to limiting TR activation during fasting (7). The NTI that occurs in response to critical illnesses could also be adaptive, as in starvation, or alternatively could be maladaptive (10). The latter interpretation originated from the robust observation that the severity of the changes in plasma thyroid hormone levels is associated with the risk of adverse outcomes, most typically mortality (11). Indeed, patients admitted to the ICU who ultimately do not survive critical illness present with much lower plasma concentrations of T4, T3, and TSH, and with higher plasma concentrations of rT3 than do survivors (11). In addition, patients suffering from prolonged critical illness present with symptoms and signs that could theoretically be eplained by a degree of hypothyroidism (12). These include unexplained impaired consciousness, suppressed myocardial function, hypothermia, diaphragm dysfunction, and failure to be weaned from the ventilator, neuropathy and weakness, kidney failure, atrophy of the skin, and hair loss. However, as these symptoms and signs could also be present due to other reasons and thus could be totally unrelated to the changes in thyroid hormone, it requires randomized controlled trials (RCTs) to address any causality in such observed associations.
Although the presence of NTI in ICU patients and the hypothesis that it could perhaps be maladaptive in this context has been repeatedly and continuously published over the last decades, the number of RCTs that address causality in association with adverse outcomes is surprisingly small (13 –16). In fact, there are only four RTCs studying a total of 190 patients that have addressed this question in adult ICU patients looking at clinically relevant outcomes (13 –16). These four very small RCTs were performed in four different study populations: patients with burn injuries, patients undergoing cardiac surgery, medical ICU patients, and patients suffering from acute kidney failure. Two of the RCTs used T4 and two used T3 as the hormone for treatment, with three out of four studies using this hormone in a pharmacologic dose. All four studies showed that the treatment further suppressed plasma TSH, and none could show a therapeutic benefit on patient outcome. With this evidence, or lack thereof, the hypothesis of NTI as a maladaptive response to critical illness cannot be confirmed. However, the hypothesis cannot be rejected with confidence either, as the studies suffered from important limitations, such as very small sample sizes and lack of statistical power. Also, the type of hormone used for treatment, the doses that were used, and the timing of treatment initiation varied. Hence, after decades and large series of mainly observational and associative studies, the question of causality still remains unanswered.
In this narrative review article, which is based on a lecture at the 2013 European Thyroid Association meeting in honor of a pioneer in the field, Prof. Georg Hennemann, several points will be made that could maybe reopen the field and identify new perspectives for future studies on the topic. The focus will be on the correct timing to identify patients at risk, given the differences between the acute and the chronic phases of critical illness, and on the role of confounders such as drugs and concomitant fasting. Without any doubt, it requires a better understanding of the pathophysiology of NTI in order to design new studies to be performed in patients at risk and to test the hypothesis of causality correctly.
NTI in Acute and Prolonged Critical Illness: Different Neuroendocrine Paradigms
Acute phase of critical illness
During acute illness, surgery, or other types of severe physical stress, there is a very rapid decline in the circulating amount of T3, whereas plasma concentrations of rT3 rise acutely (Fig. 1). These changes may at least partly be explained by reduced levels of thyroid hormone binding protein and albumin and a reduced binding activity, whereby the hormone is freed from the binding proteins and the clearance of the hormone is increased (17). Also, an acute alteration in the peripheral conversion of T4, due to a decreased D1 activity and an increased D3 activity (18,19), may explain these changes. In patients suffering from acute illnesses or other insults for which the onset can be correctly identified, such as surgery, a very transient rise in T4 has also been documented to coincide with the rapid fall in plasma T3 (20). However, in patients admitted to the ICU for reasons other than surgery, patients who are assumed to be more severely ill, admission plasma T4 concentrations are normal or at times even low (3). In addition, plasma TSH concentrations measured in a single sample can also be transiently elevated, as occurs during surgery (20), but most often plasma TSH concentrations are in the normal range in patients admitted to the ICU. However, by analyzing TSH time series, it has become clear that in this acute phase of illness, the normal nocturnal TSH surge is absent (21,22). The magnitude of the decrease in circulating T3 during the first 24 h after the onset of acute illness reflects the severity of illness and correlates with mortality (23 –26).
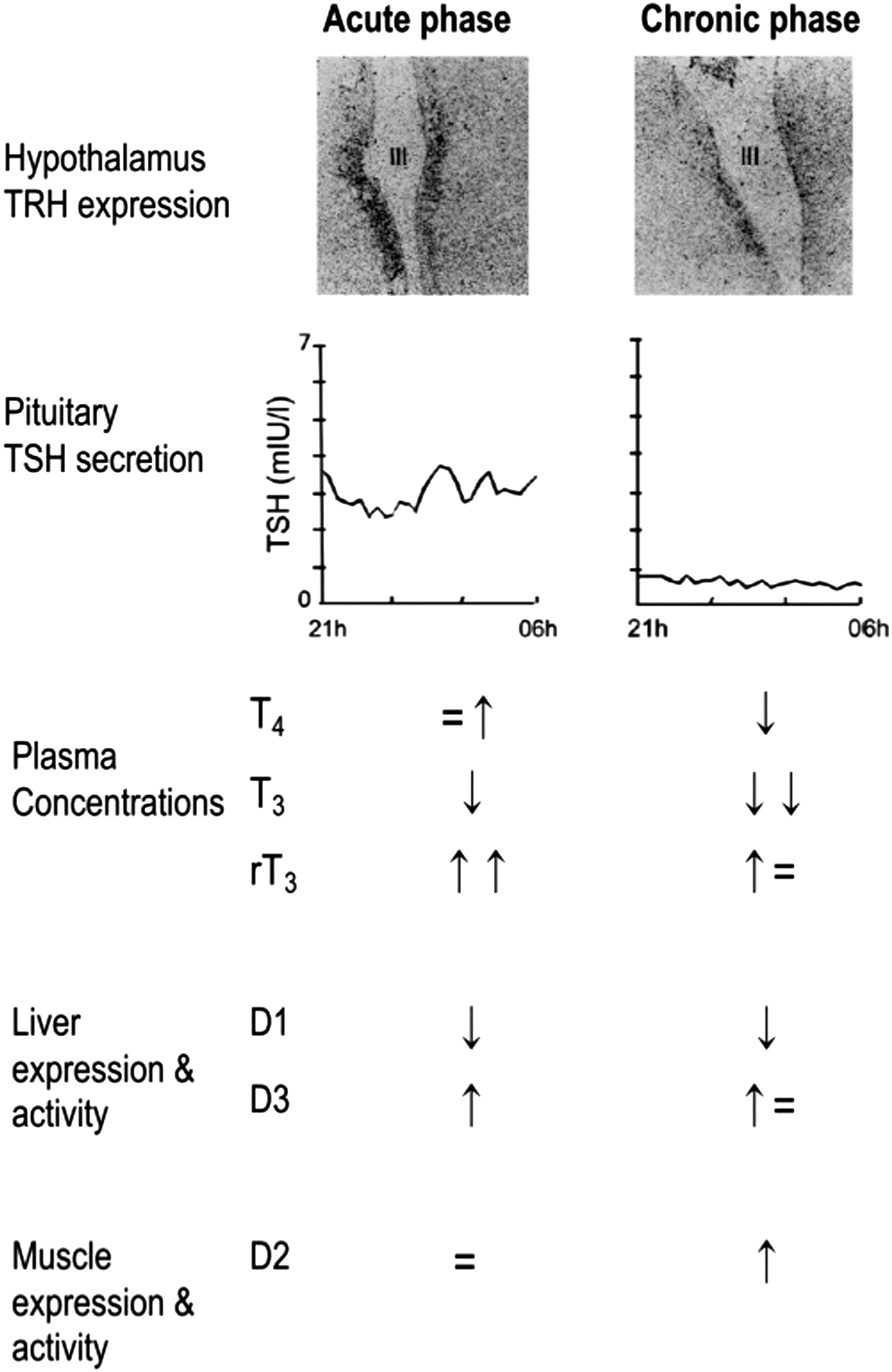
Changes in the central and peripheral thyroid axis in acute versus prolonged critical illness. The upper panel shows reduced thyrotropin releasing hormone (TRH) gene expression in the hypothalamus of prolonged ill patients. The central panel illustrates adaptations in nocturnal thyrotropin (TSH) secretion with a loss of pulsatility during prolonged critical illness. The bottom panel summarizes schematically the changes in circulating thyroid hormone concentrations and changes in peripheral deiodinase enzyme activity levels. D1, type-1 deiodinase; D2, type-2 deiodinase; D3, type-3 deiodinase. Figure reproduced, with permission, from Boonen et al. (5).
Possible mediators of the acute fall in plasma T3 concentrations in critically ill patients include the release of cytokines, metabolites, and hypoxia-induced alterations (4,27,28). The cytokines tumor necrosis factor-alpha, interleukin-1, and interleukin-6 are indeed capable of mimicking the acute illness-induced alterations observed within the thyroid axis. However, injection of neutralizing antibodies to these cytokines failed to restore normal thyroid hormone concentrations in a human experiment of lipopolysaccharide (LPS)-induced inflammation (29). Acute decreases in plasma concentrations of thyroid hormone binding proteins and albumin, and the inhibition of hormone binding, transport, and metabolism by elevated levels of free fatty acids and bilirubin may also play a role (30). A short-term intravenous infusion of T3 in patients undergoing elective cardiac surgery showed improved postoperative cardiac function (31,32). However, supranormal plasma T3 concentrations were achieved with this treatment, and thus it remains unclear whether these findings were an effect of correcting the low plasma T3 or instead merely due to a pharmacological inotropic effect of those high T3 doses.
Nutrition, or lack thereof, as a confounder during the acute phase of illness
The low T3 concentrations that occur with fasting in otherwise healthy subjects have shown to be adaptive, as they appear to protect the organism against deleterious catabolic consequences of lack of macronutrients (8,33). As patients with critical illnesses admitted to the ICU are usually unable to eat normally by mouth, and often do not receive artificial nutrition until later in the course of the illness, part of the acute fall in circulating levels of thyroid hormones in response to illness could be evoked by the concomitant fasting. Hence, a decrease in thyroid hormone availability in this context could also, at least in part, reflect an adaptive attempt to reduce energy expenditure (8). Recently, it was shown in a large RCT—the Early versus Late Parenteral Nutrition in Critically Ill Adults (EPaNIC) trial—that tolerating the pronounced macronutrient deficit that occurs in ICU patients during the first week, when only relying on enteral feeding, improves outcomes as compared with preventing that macronutrient deficit with early initiation of parenteral nutrition. Indeed, not feeding patients early reduced the incidence of nosocomial infections, reduced the occurrence of liver function abnormalities, prevented rather increased muscle weakness, and accelerated recovery (34 –37). As tolerating a fasting response in ICU patients thus appears to be beneficial, perhaps part of these adaptions are mediated via acute NTI. This hypothesis was tested in the context of the large EPaNIC randomized controlled trial (38). The changes in plasma concentrations that occurred during the first days in the ICU in that RCT in the patients randomly assigned to the “fed” or the “fasted” arm were striking: early feeding prevented a large part of the early fall in plasma TSH, T4, and T3, as well as the rise in rT3 (38) (Fig. 2). Similar effects of early parenteral nutrition, as compared with fasting during critical illness, were documented in an animal model of critical illness (39) (Fig. 3). In this study, full fasting was compared with full feeding. The early feeding not only prevented the NTI plasma concentration phenotype, but also reversed the suppressed expression and activity of liver D1 and the activated expression and activity of D3 in this animal study (39). In the clinical study, not feeding early was superior to early feeding with regard to patient outcomes, and thus this suggested that the acute fasting induced NTI during critical illness could indeed mediate some of the outcome benefit obtained with not feeding early. A complex statistical analysis indicated that part of the outcome benefit of not feeding patients early was indeed mediated specifically by the acute fall in T3 and in the T3/rT3 ratio, which indirectly supported the beneficial nature of the peripheral inactivation of thyroid hormone during acute critical illness (38). Also, these data suggest that at least part of the immediate fall in T3 concentrations during acute critical illness is related to the concomitant fasting rather than the illness per se, and that this part of the response is likely adaptive. Possible benefits of an acute NTI include the expected reduction in energy expenditure with low T3 levels, but also a direct effect of increased D3 activity locally in granulocytes, which could optimize bacterial killing capacity (4,40). The latter could in part explain the finding of fewer nosocomial infections with tolerating the macronutrient deficit during acute critical illness (34).
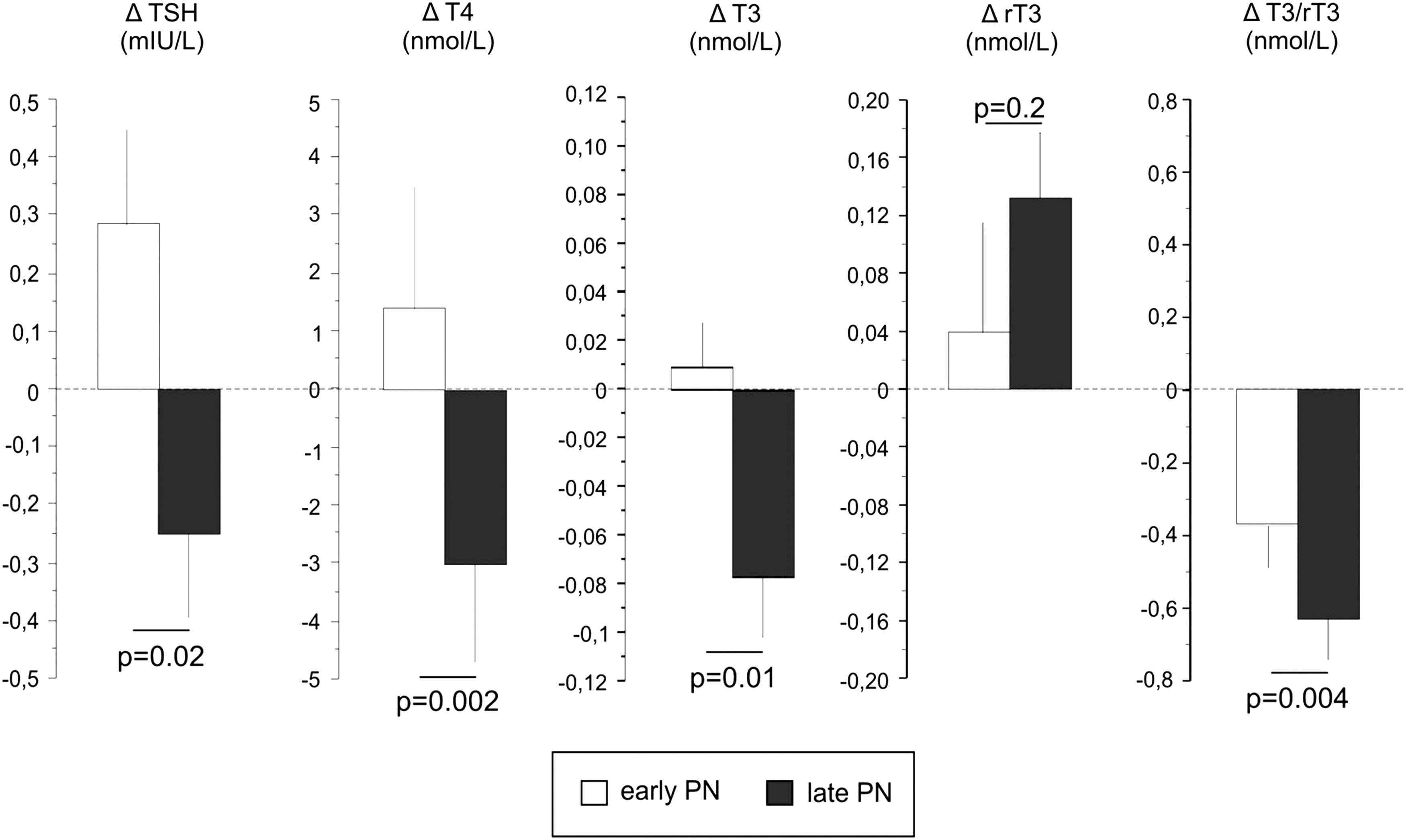
Effects of early macronutrient supplementation versus tolerating macronutrient deficit during the first three days of critical illness: partial reversal of nonthyroidal illness syndrome (NTI) in human patients. The bars (mean±standard error) represent the changes from the admission values (Δ) to day 3 in the intensive care unit (ICU; or last day for patients with a shorter ICU stay) in serum TSH, thryoxine (T4), triiodothyronine (T3), rT3, and T3/rT3. The open bars represent values of patients randomized to receiving early parenteral nutrition (early PN), whereas filled bars are presenting values from patients randomized to nutrient restriction (late PN). p-Values are those obtained with multivariable analysis, after adjustment for prespecified baseline risk factors. Figure reproduced, with permission, from Langouche et al. (38).
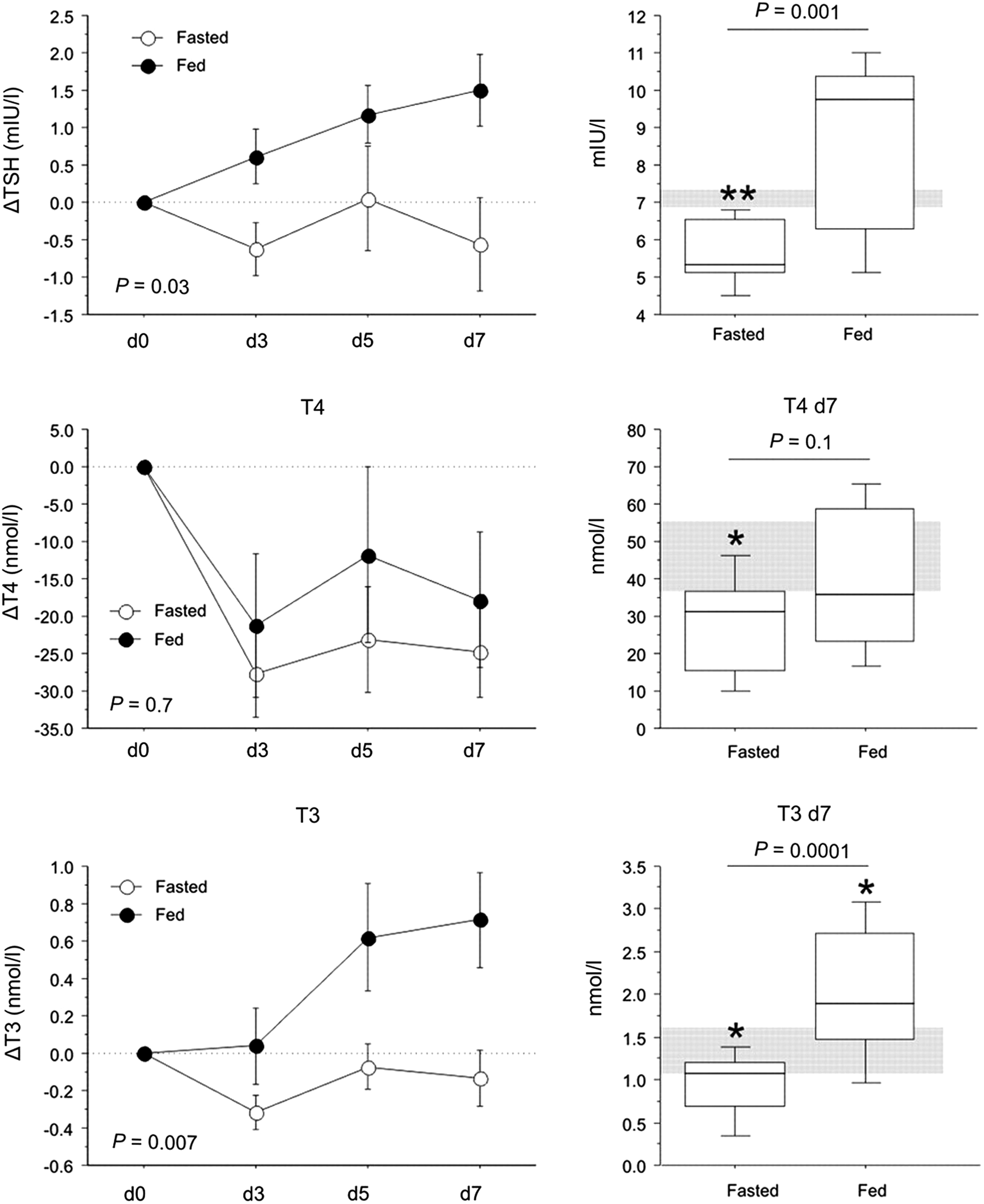
Effect of fasting versus feeding during critical illness in a rabbit model. Feeding critically ill rabbits reverses NTI compared with full fasting for 7 days after onset of illness. Figure adapted, with permission, from Mebis et al. (39).
Prolonged critical illness
However, when ICU patients are also receiving, among other vital support, full enteral and/or parenteral nutrition for several weeks or longer, the alterations within the thyroid axis appear different (3). In this phase of critical illness, low plasma T3 concentrations now coincide with low plasma T4 concentrations and low-normal TSH concentrations in a single morning sample (41). Moreover, overnight repeated sampling revealed that the pulsatility of the TSH secretory pattern is virtually lost, which relates to low plasma thyroid hormone levels, a presentation resembling central hypothyroidism (Fig. 1) (41). In line with this interpretation, Fliers et al. (42) demonstrated in postmortem brain samples of chronic critically ill patients that thyrotropin releasing hormone (TRH) gene expression in the hypothalamic paraventricular nuclei was much lower than in patients who died after acute insults (Fig. 1). Furthermore, a positive correlation was observed between the TRH mRNA expression and the plasma concentrations of TSH and T3 (42). Together, these data indicate that production and/or release of thyroid hormones is reduced in prolonged critical illness due to reduced hypothalamic stimulation of the thyrotropes, in turn leading to reduced stimulation of the thyroid gland. The observation that a rise in TSH levels precedes the onset of recovery from severe illness further supports this interpretation (43).
The factors triggering hypothalamic suppression during prolonged critical illness are unknown. Since plasma cytokine concentrations are usually much lower in the prolonged phase of critical illness (44), other mechanisms likely play role. Candidate mediators are endogenous dopamine or elevated cortisol levels in the hypothalamus, as exogenous dopamine and hydrocortisone are known to provoke or aggravate hypothyroidism in critical illness (45 –47). A local increase in type-2 deiodinase (D2) activity in the hypothalamus could elevate local thyroid hormone levels, whereby the set-point for feedback inhibition could be altered (48). Indeed, in a rabbit model of prolonged critical illness and low thyroid hormone plasma concentrations, hypothalamic TRH mRNA was low and D2 mRNA was high (49). However, the hypothalamic T4 and T3 concentrations were not increased (49). Increased pituitary D2 could also play a role in suppressing local TSH mRNA (50), although this was not confirmed in an animal model of prolonged critical illness (51).
Also, and different from what happens in the acute phase of critical illness, during prolonged critical illness, peripheral tissues seem to respond to low T3 levels by alterations that could mediate an increase in the local thyroid hormone availability and effect. For example, in skeletal muscle and liver biopsies from prolonged critically ill patients, the monocarboxylate transporter MCT-8 was overexpressed (Fig. 4) (52). This was confirmed in an animal model, where the upregulation of the monocarboxylate transporters in liver and kidney was reversable by treatment with thyroid hormones (52,53). Also, in skeletal muscle biopsies from prolonged critically ill patients, D2 expression and activity was upregulated as compared with healthy controls and with acutely ill patients (Fig. 4) (53). Upregulation of D2 in the lung was also recently found to be adaptive in sepsis and acute lung injury, further accentuated by the observation that a D2 polymorphism was found to associate with less susceptability to sepsis (54). At the level of the TR, an inverse correlation was observed between the ratio of active TRa1 over the ligand-insensitive isoform TRa2, a surrogate marker of thyroid hormone sensitivity, and the ratio of T3 over rT3 in liver biopsies of prolonged critically ill patients (55). Together, the data suggest that when the production of thyroid hormones falls in prolonged critical illness, peripheral tissues adapt by increasing thyroid hormone transporters, local activation of thyroid hormone and gene expression of the active receptor isoform.
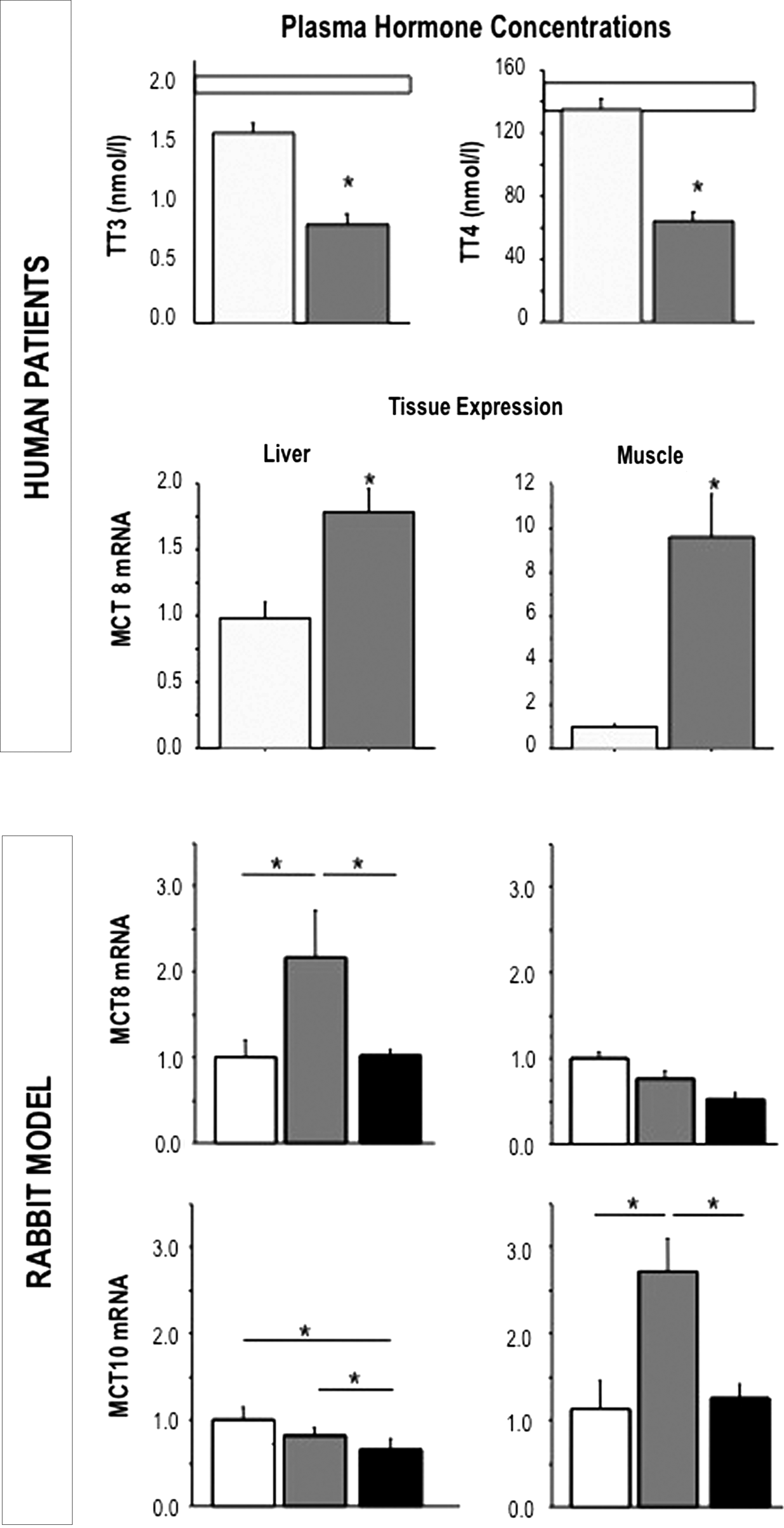
Differences between the acute and the prolonged phase of critical illness in plasma hormone concentrations and in tissue expression of the monocarboxylate transporters. The upper panel represents the circulating thyroid hormone parameters in acutely stressed (light gray bars, n=22) and chronically ill patients (dark gray bars, n=64). The white bars designate the normal ranges. The central panel shows the relative MCT8 and MCT10 mRNA expression levels measured in liver and skeletal muscle of acutely stressed (light gray) and chronically ill (dark gray) patients. The lower panels represents the relative expression levels of MCT8 and MCT10 in liver and muscle of healthy control rabbits (white bar), saline-treated prolonged ill rabbits (dark gray), and T3+T4 treated (black bar) ill rabbits. Data are expressed as mean±standard error of the mean. *p<0.05 versus acute values. Figure reproduced, with permission, from Boonen et al. (5).
In protracted critical illness, low T3 levels were found to correlate inversely with markers of muscle breakdown and of bone loss (56). Such an association could either indicate an adaptive and protective response against catabolism (low T3 as a compensatory response to hypercatabolism) or a causal maladaptive relationship (low T3 contributing to hypercatabolism) (57,58). As the cause of the low thyroid hormone levels during prolonged critical illness appears to be a suppressed TRH expression, the question could be addressed by assessing the effect of TRH treatment. When patients were given a TRH infusion, plasma T3 and T4 could be increased, but rT3 concentrations also rose (56,58). However, when TRH was combined with a growth hormone (GH)-secretagogue, this rise in rT3 was prevented (58), explained by a GH-mediated effect on the inactivating D3 (Fig. 5) (59). This treatment also induced an anabolic response that is not present with the GH-secretagogue alone, which suggests a causal relationship between low thyroid hormone levels and the impaired anabolism during prolonged critical illness (57,58). Furthermore, the negative feedback exerted by thyroid hormones on the thyrotropes was found to be maintained during TRH infusion, a self-limitation that precludes overstimulation of the thyroid axis (56,58).
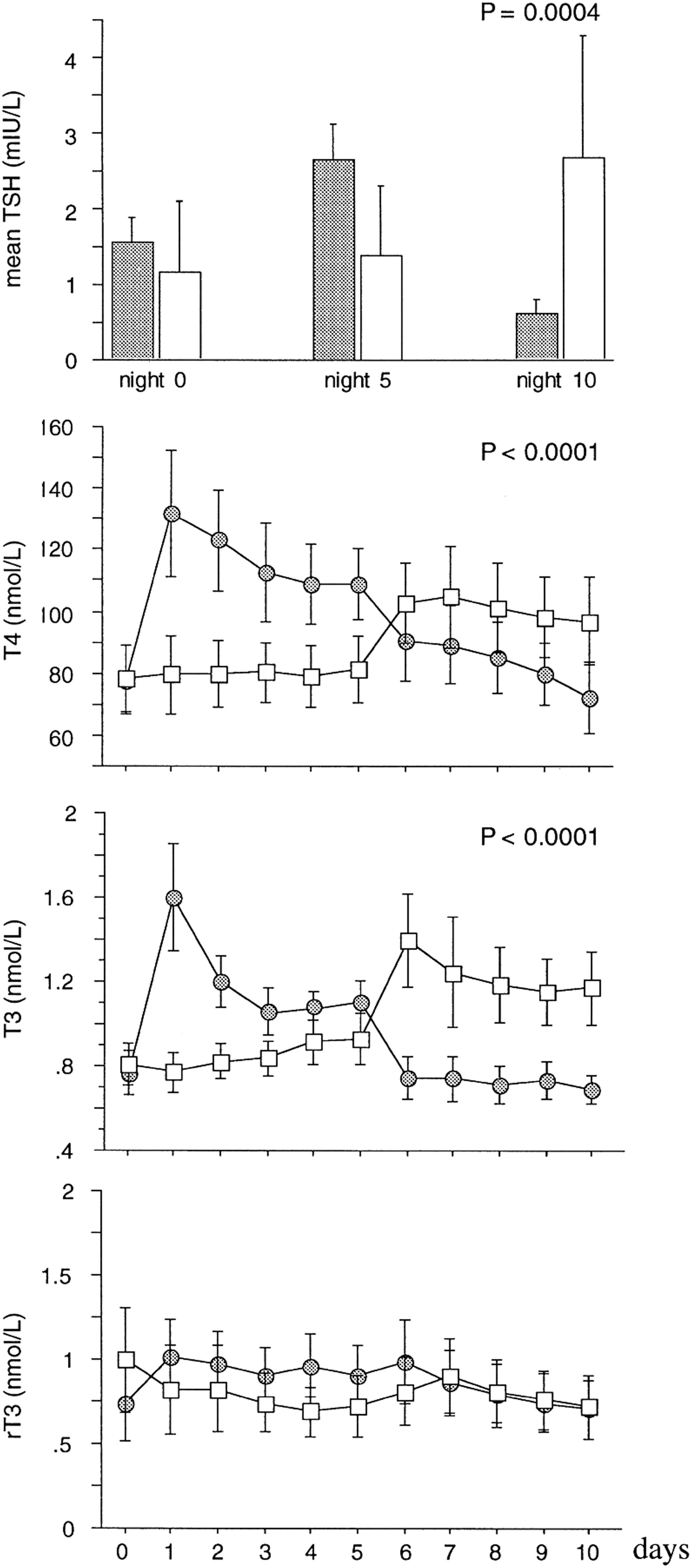
Effect of the combined infusion of TRH and a growth hormone (GH)-secretagogue on NTI during prolonged critical illness. A continuous infusion of TRH in combination with a GH-secretagogue in prolonged critically ill fed patients reversed the NTI as evidenced via a cross-over design: five days infusion of releasing peptides followed by five days placebo (filled symbols) or vice versa (open symbols). Figure reproduced, with permission, from Van den Berghe et al. (57).
Drugs as Confounders
Of course, discriminating between the acute and the chronic phase of critical illness and testing the hypothesis of causality in the association between NTI and poor outcomes should exclude all drugs that iatrogenically suppress the thyroid axis. There are many drugs that have profound effects on thyroid hormone secretion and metabolism. The reader is referred to other reviews on this topic (60), as addressing all these drug effects falls beyond the scope of this narrative review. However, one drug that has been shown to induce clinically relevant and iatrogenic hypothyroidism in critically ill adults and children is dopamine. Dopamine is a drug with vasopressor and inotrope activity that has long been the first choice to treat patients with cardiogenic or septic shock in the ICU. Even in low doses, often used in ICU patients for a considerable amount of time, dopamine has shown to profoundly suppress TSH secretion, and to lower plasma T4 and T3 concentrations in adult and pediatric ICU patients, to levels that are compatible with severe iatrogenic hypothyroidism (46,47,61) (Fig. 6). Indeed, upon withdrawal of the dopamine infusion, the release of TSH was found to rebound, which brought about a substantial and sustained rise in T4 and T3 (46,47,61). Treating pediatric ICU patients who are on a dopamine infusion with thyroid hormone was shown to improve intensive care outcome in a RCT (62). Clearly, in order to address whether the illness-induced alterations in the thyroid axis require treatment, such iatrogenic confounders by drugs in the ICU should be excluded.
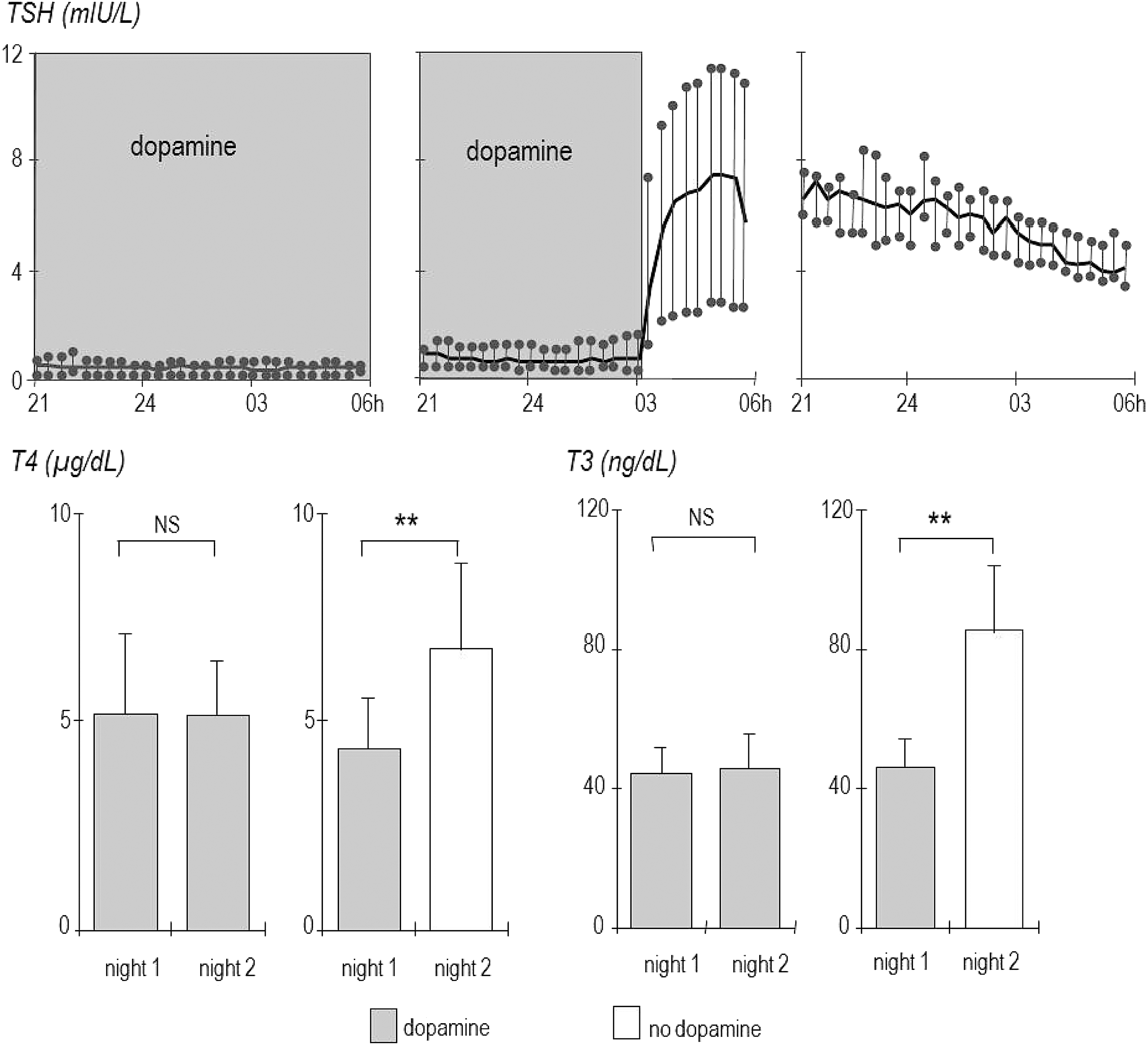
The effect of dopamine infusion/withdrawal on plasma TSH concentrations and on plasma T4 and T3 concentrations. Upper panels: during dopamine infusion, TSH levels are suppressed, whereas within 20 minutes of withdrawal, TSH release shows a rebound, with higher levels the day after dopamine withdrawal. Lower panels: 24 hours after dopamine withdrawal, plasma T4 and T3 concentrations are much higher than during dopamine infusion. Figure adapted, with permission, from Van den Berghe et al. (46).
Which Patients and Which Hormone for Future Studies?
As NTI occurring in the acute phase of critical illness is to a large extent explained by concomitant fasting, and as acute NTI likely brings about adaptive responses that are selected by evolution, the acute phase of critical illness is not a condition in which the impact of thyroid hormone should be further investigated. Indeed, there is not a good rationale for any benefit of such treatment. In contrast, patients in the prolonged phase of critical illness, who are fully fed via the enteral or the parenteral route, who present with low plasma T4 and T3 concentrations, and who show symptoms and signs that could be compatible with (non-iatrogenic) hypothyroidism may be different. Perhaps these patients should be considered as patients “at risk” and thus could be eligible for inclusion in RCTs to assess the effect of treatment. Indeed, in these patients, the cause of NTI lies in the suppressed hypothalamic TRH expression, and thus NTI in these patients could be a form of central hypothyroidism. Also, treatment of patients in ICUs for weeks or months is a recent phenomenon in the evolution of mankind, and thus any alterations that occur in this unnatural phase of the stress response should not per se be interpreted as beneficial and “selected” by evolution. Finally, the peripheral responses to the low T3 levels appear to indicate compensation and attempts to increase thyroid hormone availability.
What would be the hormone of choice for such future studies? In a set of animal studies, the impact of treatment with T4, T3, or the combination of T4 and T3 has been investigated and compared with treatment with TRH (63 –65). These studies showed that in prolonged critical illness, substitution doses of T4 or T3 are unable to bring about a rise in plasma concentrations of thyroid hormones, whereas an infusion of TRH was able to increase T4 and T3 (63 –65). A supraphysiologic dose of T4, which is able to normalize plasma T3 during critical illness, resulted in far too high plasma T4 concentrations and suppressed TSH. Also a supraphysiologic dose of T3 that allowed a normalization of plasma T3 suppressed plasma TSH and T4 to subnormal levels (63 –65). It is clear, too, from old clinical studies that it is very difficult to normalize plasma thyroid hormone concentrations by using T4 and/or T3 in the context of critical illness. As the cause of the problem in the prolonged phase of critical illness is not the thyroidal capacity to produce or release thyroid hormone, nor the peripheral metabolism or uptake, but instead lies in a suppressed hypothalamic expression of releasing factors, such as TRH but also GH-releasing hormones, maybe such peptides are a better and a more rational alternative. Indeed, in a set of clinical studies, the combination of TRH and GH-releasing peptide-2 has been shown to reactivate the thyroid axis without increasing rT3 and to evoke an anabolic reponse at the tissue level, confirming that such combinations of releasing factors may be more rational and safer to test in future RCTs than T4 and/or T3.
Conclusions
It is clear that the name “NTI” during critical illness refers to a syndrome with different faces. Some of it is good and some of it may be bad, depending on the timing and the context. The acute stress or critical illness-induced alterations within the thyroid axis as these occur in the first days of critical illness are brought about at least in part by the concomitant macronutrient deficit. Tolerating this early “fasting response” and its concomitant changes in thyroid hormone parameters appears to be wise and beneficial. However, the “NTI” that occurs in prolonged critically ill patients, who continue to be dependent on intensive medical care for weeks or months, may have a different face, both in its origin and in its impact on outcome. Future studies should perhaps target this specific and selected patient population at risk, after excluding iatrogenic drug interferences, and investigate the effect on outcome of treatment with hypothalamic releasing factors in adequately powered RCTs.
Footnotes
Acknowledgments
The work summarized in this review has been supported by research grants from the Fund for Scientific Research Flanders Belgium, the Methusalem Program funded by the Flemish Government, and the European Research Council under the European Union's Seventh Framework Program (FP7/2007–2013 ERC Advanced Grant Agreement no. 307523).
Author Disclosure Statement
The author has no conflict of interest to declare.