
Abstract
Background:
Congenital hyperthyroidism can be a cause of failure to thrive, hyperactivity, developmental delay, and craniosynostosis during infancy. Most commonly, the condition occurs in the setting of maternal autoimmune thyroid disease. Rarely, congenital hyperthyroidism can also occur secondary to activating mutations within the thyrotropin (TSH) receptor.
Patient Findings:
A Hispanic male infant presented at age 6 months with severe thyrotoxicosis. At the time of presentation he was being evaluated for squamosal suture synostosis and he was noted to have significant developmental delays.
Summary:
The patient's thyrotoxicosis was initially treated with antithyroid medication, and he subsequently underwent craniosynostosis repair leading to neurodevelopmental improvement. DNA from the patient and his parents was submitted for mutational analysis of exons 9 and 10 of the TSH receptor. He was found to carry a monoallelic transition 1895C>T in exon 10 that resulted in the substitution of threonine at position 632 by isoleucine (T32I). This mutation resulted in constitutive activation of the TSH receptor. Neither parent carried this mutation indicating that the child has acquired a de novo germline mutation.
Conclusions:
We report the first case of squamosal suture craniosynostosis in a patient with non-autoimmune hyperthyroidism. Squamosal suture craniosynotosis is rare, often has a subtle presentation, and should be considered in all patients with this condition because prompt treatment of hyperthyroidism and craniosynotosis repair can lead to normalization of neurodevelopment.
Introduction
R
Patient
The patient was a boy born at 35 5/7 weeks. At age 5 months, he was diagnosed with bilateral squamosal suture craniosynostosis (Fig. 1). At age 6 months, given FTT and a history of craniosynostosis, his pediatrician obtained thyroid function tests notable for a free thyroxine (FT4) level of 7.2 ng/dL (reference: 0.89–1.6 ng/dL) and a TSH level of <0.01 mIU/L. That evening he developed a rash and fever reaching 40°C. He was admitted with a diagnosis of severe hyperthyroidism and concern for impending thyroid storm. There was no known family history of thyroid disease or autoimmune disorders.
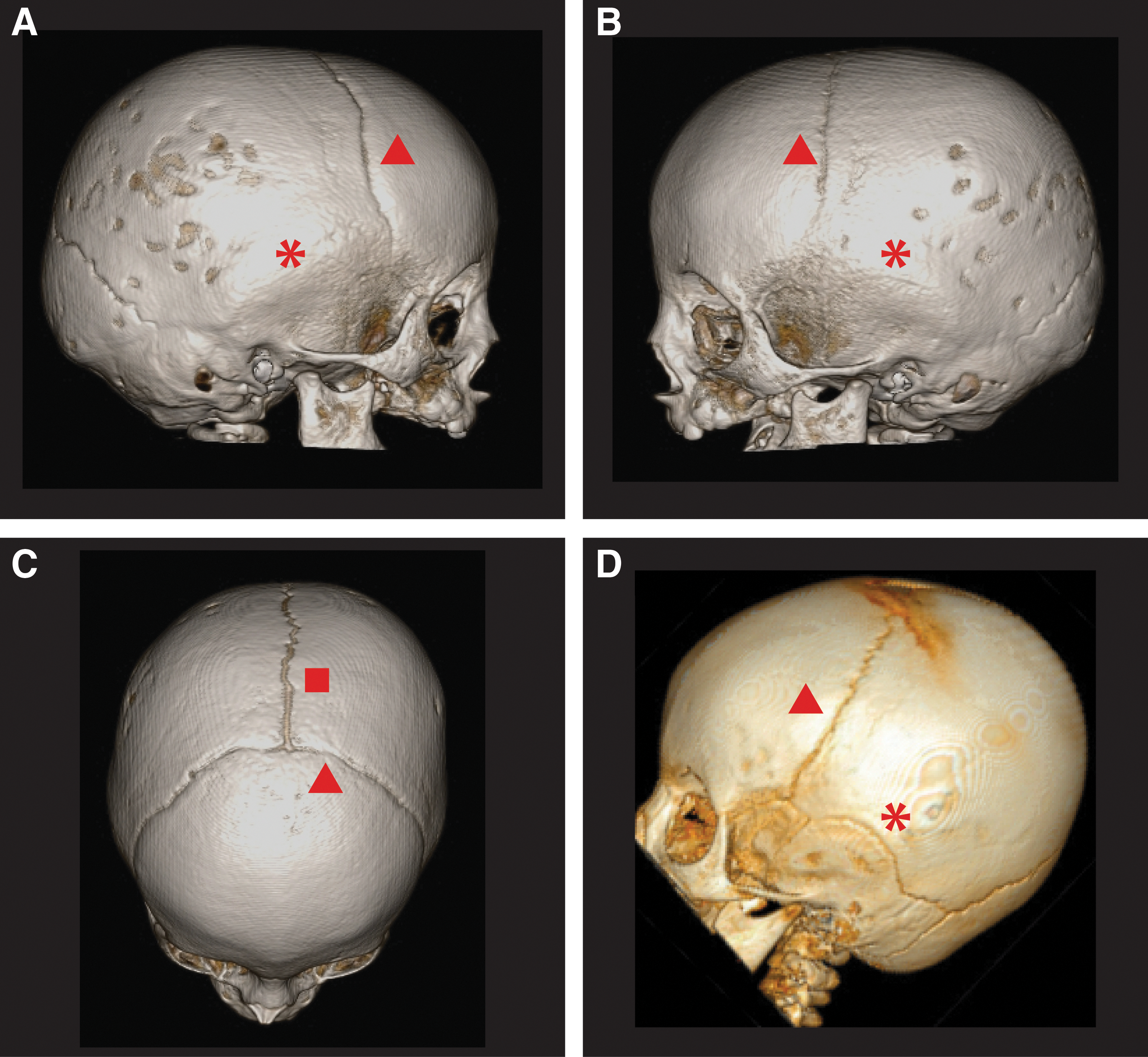
Three-dimensional computed tomography images of the cranium and associated sutures of the patient
On admission, the patient was tachycardic, with a heart rate at 180 beats per minute, and febrile, with a temperature of 38.4°C. His weight was 7 kg (14th percentile), length was 71 cm (90th percentile), and his head circumference was 42.1 cm (7th percentile). Physical exam revealed plagiocephaly (i.e., flattening of one side of the skull), closed fontanelles, a nonpalpable thyroid, and developmental delays. He was unable to roll over, unable to sit unsupported, and he was not babbling. He also had difficulty lifting his head completely when prone, which was a regression noted by his parents in the previous 2 months. Repeat thyroid studies were significant for total triiodothyronine (TT3) at 385 ng/dL (reference: 79–272 ng/dL), FT4 at 4.5 ng/dL (0.89–1.6 ng/dL), and TSH at <0.01 mIU/L (reference: 0.4–8.2 mIU/L) (Table 1). He had a mild elevation in liver enzymes with an alanine aminotransferase (ALT) level of 81 IU/L (3–41 IU/L) and an aspartate aminotransferase (AST) level of 68 IU/L (15–59 IU/L) (Table 1). He was treated for presumptive thyroid storm with methimazole, hydrocortisone, propranolol, and SSKI (saturated solution of potassium iodide). No further fevers occurred, hydrocortisone was gradually stopped within 24 hours, and SSKI was gradually stopped 3 days later. A thyroid ultrasound demonstrated homogenous parenchyma and no enlargement. The right lobe measured 1.1×1.9×1.3 cm with a volume of 1.28 mL, and the left lobe measured 1.1×1.8×1.2 cm with a volume of 1.6 mL. His bone age was advanced at 4.4 years using the Tanner and Whitehouse (TW2) method. Thyroid-stimulating immunoglobulin (TSI) levels and antithyroglobulin antibodies were not elevated (TSI <89% [normal <140%]; antithyroglobulin antibody <280 IU/mL [normal <280]). Parental thyroid function tests were normal, and no elevation in maternal TSI was noted. His thyroid function improved with treatment, and he was discharged 5 days after admission on therapy with 1.25 mg of methimazole three times daily and 1.75 mg of propranolol three times daily.
ALT reference range: 3–44 IU/L, then 3–34 IU/L due to a change in the lab assay. AST reference range: 15–54 IU/L, then 22–59 IU/L, then 24–65 IU/L due to a change in the lab assay. Age-adjusted FT4 reference range: 1–11 months (0.89–1.6 ng/dL), 1 year (0.87–1.5 ng/dL), and 2–17 years (0.96–1.77 ng/dL). Age-adjusted TT3 reference range: 1–11 months (79–272 ng/dL), 1 year (62–272 ng/dL), and 2–17 years (92–248 ng/dL). Age-adjusted TSH reference range: 1–11 months (0.4–8.2 mIU/L), 1 year (0.4–5.7 mIU/L), and 2–17 years (0.7–5.97 mIU/L).
ALT, alanine aminotransferase; AST, aspartate aminotransferase; MMI, methimazole; FT4, free thyroxine; TSH, thyrotropin; TT3, total triiodothyronine.
At his follow-up visit 1 week later, he was again rolling back to front, was able to hold his neck up and turn his head, and had begun to babble. He was also “less jittery” while sleeping, and he had gained 0.6 kg (1 lb, 5 oz). His FT4 and TT3 had normalized with a level of 1.41 ng/dL (reference: 0.89–1.6 ng/dL) and 200 ng/dL (79–272 ng/dL), respectively (Table 1). Additionally, ALT had nearly normalized at 47 IU/L (reference: 3–44 IU/L), and AST had normalized at 39 IU/L (15–54 IU/L) (Table 1). When rechecked nearly 3 weeks later, both AST and ALT had fully normalized with ALT at 21 IU/L and AST at 34 IU/L, suggesting that the initial mild transaminitis was due to uncontrolled hyperthyroidism. The propranolol was discontinued, and the patient remained euthyroid on methimazole 1.25 mg twice daily.
He successfully underwent craniosynostosis repair at age 9 months. At age 12 months, while on 1.25 mg twice daily of methimazole, he presented again with overt hyperthyroidism: his FT4 level was 6.19 ng/dL (reference: 0.87–1.5 ng/dL) and his TT3 level was 590 ng/dL (62–272 ng/dL) (Table 1). The liver enzymes were also elevated: ALT, 156 IU/L (3–34 IU/L) and AST, 82 IU/L (22–59 IU/L). Methimazole was thought to be contributing to the abnormal liver function, which nearly normalized after its discontinuation: ALT, 39 IU/L and AST, 37 IU/L (Table 1). The patient successfully underwent thyroidectomy at age 13 months. His postoperative course was complicated by hypocalcemia, and he was discharged on calcitriol, elemental calcium, and levothyroxine replacement at 2.5 μg/kg/d. During this time, motor development continued to proceed normally.
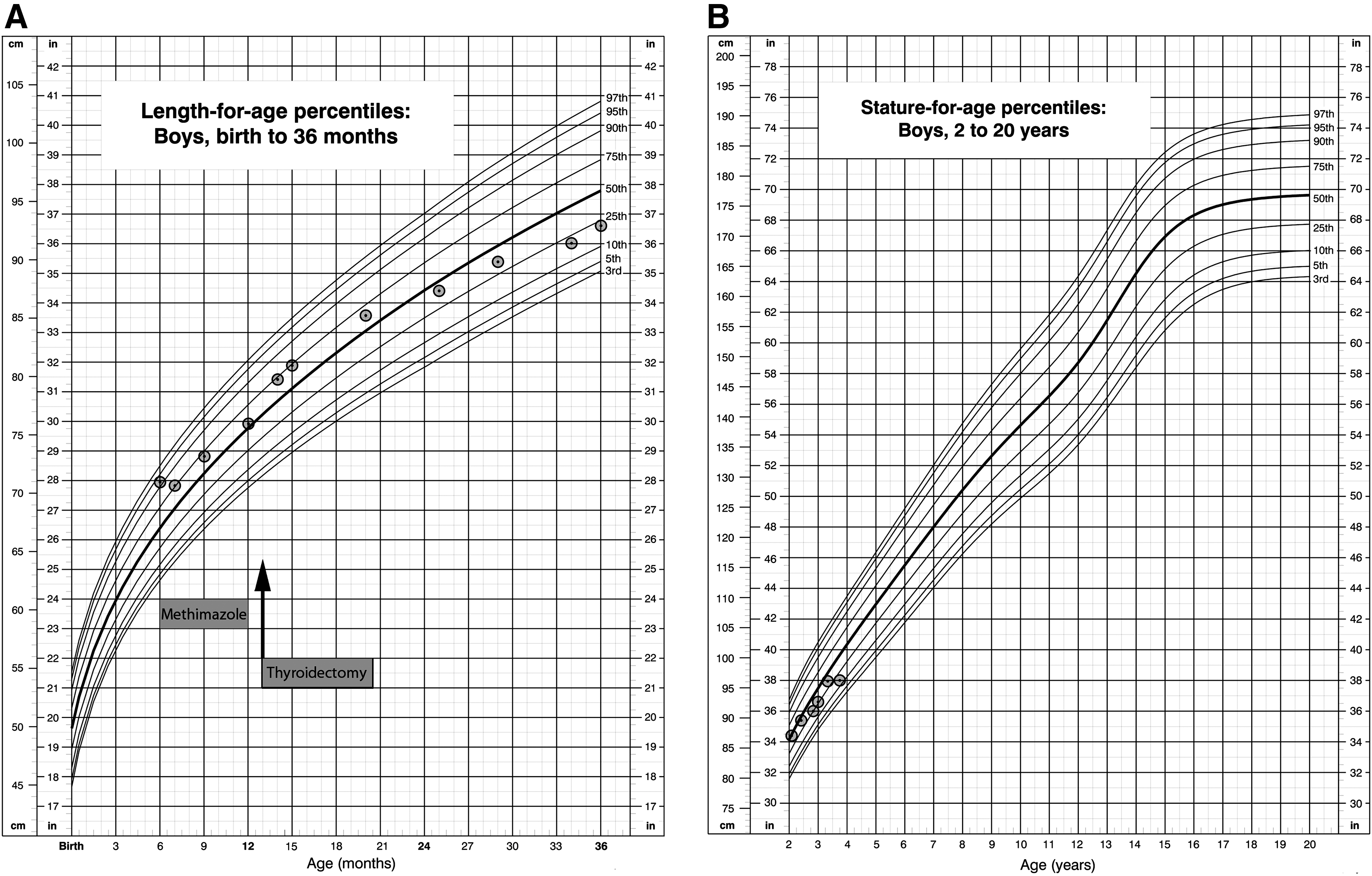
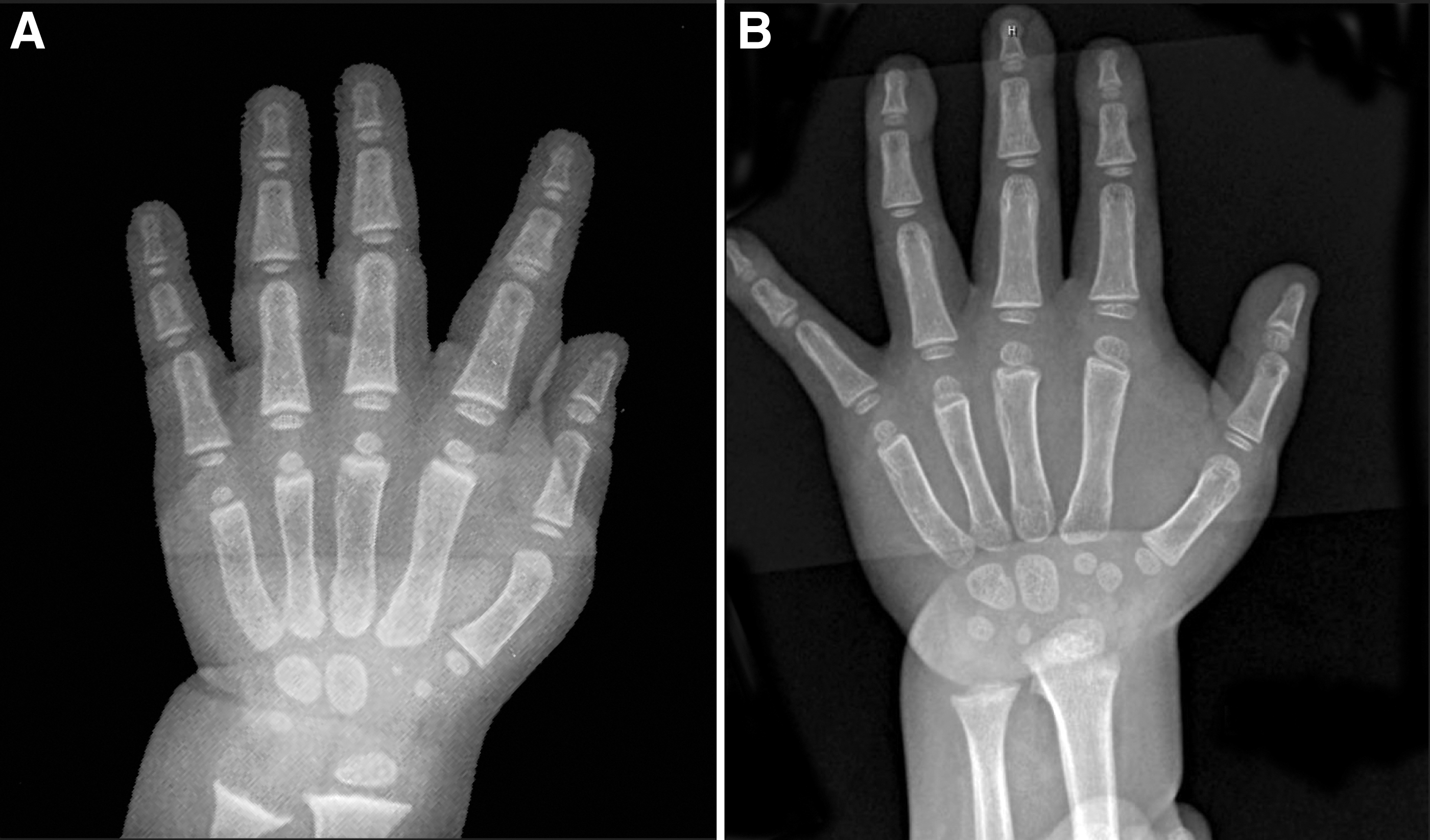
From age 13 months onward, once euthyroid, rapid bone advancement ceased. At age 14 months, his bone age was read as 4.4–4.5 years (RUS TW2) and at age 20 months his bone age was read as 4.6–4.7 years (Fig. 2). As expected, his linear growth velocity also decreased. The growth velocity decreased from 10 cm/y at 12 months (height of 76 cm, 56th percentile) to 0.6 cm/y (height of 18th percentile) at 45 months (Fig. 3).
Bone age films of the index patient.
Linear growth charts (U.S. Centers for Disease Control and Prevention) for the index patient.
Currently, at age 45 months, he is euthyroid on levothyroxine 3 μg/kg/d and remains on calcitriol 0.2 μg daily and elemental calcium 22 mg/kg/d. His weight is 13.8 kg (10th percentile) and his height is 96.6 cm (18th percentile). There has been no further advancement in bone age. He is walking and running well with normal gross motor development. Early Intervention assessment at age 25 months was notable for delays in expressive and receptive language (functioning at age 12–18 months) and temper tantrums for which he received speech and behavioral therapy. He has now completed these therapies and is developing age appropriately.
Mutational Analysis
Because of the absence of evidence for an autoimmune etiology, the TSH receptor gene was submitted for mutational analysis. After obtaining informed consent, DNA was extracted from peripheral blood from the patient and his parents. Exons 9 and 10 were amplified by polymerase chain reaction and sequenced. Sequence analysis revealed a monoallelic transition 1895C>T in exon 10 resulting in the substitution of threonine 632 by isoleucine (T632I) in the sixth transmembrane of the receptor. This mutation has been previously reported (8 –11) and results in constitutive activation of the TSH receptor. Neither parent carries this missense mutation, suggesting that the patient had a de novo germline mutation.
In addition, the patient and his father both possess a monoallelic single nucleotide polymorphism 2181C>G in exon 10 that results in replacement of aspartic acid 727 with glutamic acid (D727E). Although this allele has been associated with toxic multinodular goiter (12), it does not seem to result in altered function of the TSH receptor (13).
Discussion
Although the spectrum of long-term neurologic sequelae of hyperthyroidism during infancy is not well established, many patients with NAH have been reported to have varying degrees of mental retardation, speech disturbance, and hyperactivity (4,5). Additionally, remnant reactivation is common even after near-total thyroidectomy, necessitating treatment with radioactive iodine. Although the patient reported here is now developing age appropriately, he did require speech and behavioral therapy, and his long-term developmental outcome could potentially be influenced by hyper- or hypothyroidism.
Importantly, prior to the diagnosis of thyrotoxicosis, this patient was being evaluated for craniosynostosis (Fig. 1), a known complication of neonatal, infantile, and juvenile hyperthyroidism. In euthyroid full-term infants, the posterior fontanelle typically is not palpable beyond 2 months of age, and the anterior fontanelle is no longer palpable between 10 and 24 months of age. The metopic suture begins to fuse at approximately 2 months of age, the sagittal suture begins to fuse at approximately 22 months of age, coronal suture fusion begins at 24 months of age, and lambdoid suture fusion begins at 26 months of age (14). When multiple sutures are involved, increased intracranial pressure, cranial deformity, and mental retardation can occur, necessitating early surgical repair. Excess thyroid hormone primarily leads to premature fusion of the sagittal and coronal sutures (15,16). Akita et al. (16) demonstrated that hyperthyroidism leads to preferential early narrowing of the sagittal suture in infantile rats. Furthermore, sagittal suture synostosis has been described in a patient with the same mutation as reported here (8), but as illustrated by our patient, hyperthyroidism can be associated with bilateral complete squamosal suture synostosis. Squamosal suture synostosis is rare—in fact this suture typically does not completely fuse until adulthood. Patients with premature squamosal synostosis often present with features of occipital flattening, shortened biparietal diameter, and bossing of the parietal region (17). These findings can be subtle, but when found in association with other midline calvarial suture synostosis, the phenotype can be more pronounced (17).
Ossification is highly regulated by thyroid hormone. T3 regulates chondrocyte differentiation, enhances cartilage matrix mineralization, and directly stimulates osteoblast cell differentiation and bone matrix synthesis and degradation (18). During endochondral ossification, mesenchymal stem cells differentiate into chondrocytes that secrete cartilage matrix followed by osteoblast invasion and mineralization (19). Expectedly, this process is accelerated in the setting of hyperthyroidism, leading to bone age advancement. In contrast, intramembranous ossification of cranial sutures involves direct differentiation of mesenchymal cells into osteoblasts (19). It is therefore conceivable that differential collagen production or alteration of T3-induced osteoblast differentiation and bone matrix synthesis may have led to the preferential fusion of the squamosal suture in our patient.
Alternatively, the predisposition of thyrotoxicosis to sagittal and coronal suture synostosis may be explained by differences in embryological development. The neurocranium primarily arises from mesoderm-derived structures; however, the squamosal suture is derived from the neural crest component of the temporal bone (17). Perhaps decreased sensitivity of neural crest tissue to the effects of T3 may usually lead to sparing of the squamosal suture; alternatively the subtle features of squamosal craniosynostosis may not be readily recognized, resulting in an underestimation of its prevalence in infants with hyperthyroidism.
In addition to thyroid hormone, TSH has also been postulated to exert a direct influence on bone. The TSH receptor is expressed in both osteoclasts and osteoblasts, suggesting that TSH may have a direct role in bone metabolism (20). In several instances, TSH has been shown to reduce osteoclastic bone resorption and increase bone volume (20 –22). In rodents, low doses of TSH increased bone volume and improved bone microarchitecture independent of an increase in serum thyroid hormone levels (23,24). However, despite constitutive activation of the mutant TSH receptor, bone age advancement and linear growth acceleration were halted after thyroidectomy in the patient reported here, arguing against a major role for TSH in the regulation of bone maturation and growth.
In conclusion, this patient with NAH caused by a T632I activating mutation in the TSH receptor presented with FTT, development delay, and complete synostosis of the squamosal sutures, an unusual form of craniosynostosis. Prompt recognition and adequate treatment of hyperthyroidism and craniosynostosis are essential for normal neurodevelopment.
Footnotes
Author Disclosure Statement
The authors have nothing to disclose.