
Abstract
Background:
Population-based studies have demonstrated an association of single nucleotide polymorphisms close to the thyroid transcription factor forkhead box E1 (FOXE1) gene with thyroid cancer. The dysregulation of forkhead proteins is increasingly recognized to play a role in the development and progression of cancer. The objective of the study was to seek to identify novel mutations in FOXE1 in papillary thyroid cancer (PTC) and to assess the effect of these mutations on protein expression and transcriptional function on FOXE1 responsive promoters.
Methods:
The study was conducted at two tertiary referral hospitals. The coding region of FOXE1 was sequenced in tissue-derived DNA or RNA from 120 patients with PTC and 110 patients with multinodular goiter (MNG). In vitro studies were performed to examine the protein expression and transcriptional function of FOXE1 mutants. A molecular model of the forkhead domain (FHD) of FOXE1 was generated using the SWISS-MODEL online server with the three-dimensional structure of FOXD3 as a template.
Results:
Three somatic missense mutations were detected in PTC resulting in the amino acid substitutions P54Q, K95Q, and L112F. One additional mutation was detected in a MNG (G140R). In vitro studies demonstrated marked impairment in transcriptional activation by all four FOXE1 mutants, which was not explained by differences in protein expression. Molecular modeling localized three of the mutations to highly conserved regions of the FHD.
Conclusions:
We have identified novel somatic mutations of FOXE1 in PTC. Mutational inactivation of FOXE1 is an uncommon event in thyroid tumors but may contribute to thyroid carcinogenesis and dedifferentiation in concert with other oncogenic drivers.
Introduction
P
FOXE1 is a member of the forkhead family of transcription factors. These factors share a highly conserved winged-helix or forkhead DNA binding domain (FHD). Along with the other thyroid-specific transcription factors NK2 homeobox-1 (NKX2-1) and paired box 8 (PAX8), FOXE1 plays a critical role in normal thyroid development (8). Germline inactivating mutations in the FHD of FOXE1 result in Bamforth-Lazarus syndrome (OMIM No. 241850) which is characterized by congenital hypothyroidism due to thyroid agenesis, cleft palate, choanal atresia, and spiky hair (9).
The dysregulation of FOX family transcription factors is increasingly recognized to play a role in the development and progression of cancer. A somatic mutation in forkhead box L2 (FOXL2) is pathognomonic of adult granulosa cell tumors of the ovary (10). Mutations in forkhead box O1 (FOXO1) occur in a subset of diffuse large B-cell lymphomas and have prognostic significance (11). Forkhead box M1 (FOXM1) overexpression has been observed in a variety of tumors with effects on cell proliferation and genomic instability (12).
We hypothesized that somatic mutations in FOXE1 may be present in thyroid tumors with functional significance. We sequenced panels of PTC and benign thyroid tissue for mutations in FOXE1 to identify four independent missense mutations.
Materials and Methods
Tumor tissues
Study samples consisted of two cohorts of patients with PTC and multinodular goiter (MNG). Samples from the first cohort were obtained through the Victorian Cancer Biobank and included 50 tumor RNAs and 10 MNG RNAs derived from frozen tissue. Samples from the second cohort were from the Neuroendocrine Tumor Bank located at the Kolling Institute, Royal North Shore Hospital, and included 70 tumor DNAs and 100 MNG DNAs derived from frozen tissue. For patients in whom a mutation was identified, DNA derived from peripheral blood leukocytes was used for sequencing where available. All patients gave written informed consent for the collection of tissue and clinical data.
RNA and DNA extraction
RNA was extracted from frozen tissue using the RNeasy Mini kit (QIAGEN). DNA was extracted from frozen tissue using the Puregene extraction kit (QIAGEN) and from serum using the QIAamp DNA Mini kit (QIAGEN).
RT-PCR and sequencing
Total RNA (1 μg) was reverse-transcribed for 60 min at 50°C in a total volume of 20 μL using SuperScript III reverse transcriptase (Invitrogen). First-strand synthesis was performed using 250 ng random hexamers. The entire coding region of FOXE1 was sequenced in PTC and MNG to identify any mutations. Polyalanine allele genotypes were determined in all but one of the PTC and MNG and the minor allele frequency (FOXE116Ala
or FOXE117Ala
) in the two cohorts was compared. PTC were also tested for BRAF mutations as described previously (13). Details of the primer pairs, polymerase chain reaction (PCR) conditions, and sequencing protocol can be found in the Supplementary Methods (Supplementary Data are available online at
Transient transfection and reporter assays
An expression vector (pcDNA3.1) containing wild type FOXE1 cDNA (7) was used as the template for PCR-mediated site-directed mutagenesis to generate the four FOXE1mutant plasmids (FOXE1P54Q, FOXE1K95Q, FOXE1L112F, and FOXE1G140R); constructs were verified by restriction digest and Sanger sequencing. The transcriptional activity of the FOXE1 variants was determined using two FOXE1-responsive luciferase reporter genes, the human thyroperoxidase promoter (TPO-LUC), and an artificial construct (Z16TK-LUC) (14). An additional construct containing a mutation previously associated with Bamforth-Lazarus syndrome (FOXE1A65V) (14) was included as a positive control for transcriptional impairment. Transfection experiments were carried out in Nthy-ori-3.1 cells, a cell line derived from normal human thyroid follicular epithelial cells which has been shown to retain features of thyroid-specific differentiation (15). Cells were plated at 2×105 cells/well in a 24-well plate 24 h prior to transfection. They were then transiently cotransfected using Xtreme Gene HP transfection reagent (Roche) with 500 ng of firefly reporter, 50 ng PRL-SV40 Renilla reporter, and 100 ng of empty pcDNA3.1, pcDNA 3.1-FOXE1wild type, or pcDNA3.1-FOXE1mutant. After 24 h, cells were lysed and assayed for luciferase activity with the Dual Luciferase Reporter Assay (Promega).
As several previous studies that have examined the effect of germline FOXE1 mutations on transcriptional activity were conducted in human embryonic kidney (HEK) 293 cells (14,16,17), further transfection experiments were carried out in this cell line. The transcriptional activity of three FOXE1 mutant plasmids (FOXE1P54Q, FOXE1L112F, FOXE1G140R) was determined using the Z16TK-LUC promoter. FOXE1A65V was again used a positive control for transcriptional impairment. Further details can be found in the Supplementary Methods.
Western blot
To compare the relative expression of wild type and mutant FOXE1 proteins, NThy-ori-3.1 cells were transiently transfected with the relative expression plasmid construct, incubated for 24 h, and then lysed in ice-cold radio-immunoprecipitation assay cell lysis buffer. The protein concentration of each sample was determined using the RC DC Protein Assay kit (Bio-Rad) and then 20 μg of each protein sample was resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis and electroblotted onto Hybond ECL membrane (GE Healthcare Life Sciences). The membrane was incubated overnight at 4°C with a 1:1000 dilution of anti-FOXE1 rabbit monoclonal antibody (Abcam), and to confirm equal protein loading, a 1:10,000 dilution of anti–glyceraldehyde 3-phosphate dehydrogenase rabbit monoclonal antibody (Cell Signalling Technology). The blot was then incubated for 1 h at room temperature with a 1:10,000 dilution of goat anti-rabbit immunoglobin-G horseradish peroxidase–linked antibody (Cell Signalling) and developed using the ECL-Prime Western Blotting Detection Reagent (GE Healthcare Life Sciences). Densitometric analysis of western blots was performed using a multigauge imaging system (FujiFilm).
In silico analysis and molecular modeling
An in silico analysis tool, Polyphen-2 (
Results
The entire coding region of FOXE1 was sequenced in 120 PTC and 110 MNG. Two previously recognized synonymous SNPs (c.1047T>C and c.1485C>T) were common in both groups. Polyalanine allele frequencies (Supplementary Table S1) were significantly different in PTC compared with MNG cohorts (p<0.0001; odds ratio 4.51 [95% confidence interval: 2.57–7.90] for carriage of FOXE116Ala or FOXE117Ala alleles).
Three missense mutations were detected in PTC: c.821C>A, p.P54Q; c.943A>C p.K95Q; and c.994C>T, p.L112F. Each mutation occurred in a single tumor, and two of the mutations (p.P54Q and p.L112F) were detected in tissue derived from recurrent tumors. One missense mutation was detected in a single MNG: c.1078G>C, p.G140R. In two of the three PTC and in the MNG, FOXE1 was sequenced in both RNA and DNA extracted from the tumor/tissue, confirming a mutation rather than an RNA editing event.
All four mutations occurred in the FHD of FOXE1 (Fig. 1) and were predicted to be damaging by PolyPhen-2 (Supplementary Fig. S1). The clinical characteristics and BRAF mutation status of the mutant FOXE1 PTC patients are listed in Table 1. p.P54Q, p.K95Q, and p.G140R were heterozygous mutations, whereas p.L112F was hemizygous; loss of allelic expression in tumor RNA from this latter patient was demonstrated for two downstream SNPs in FOXE1 (Supplementary Fig. S2).
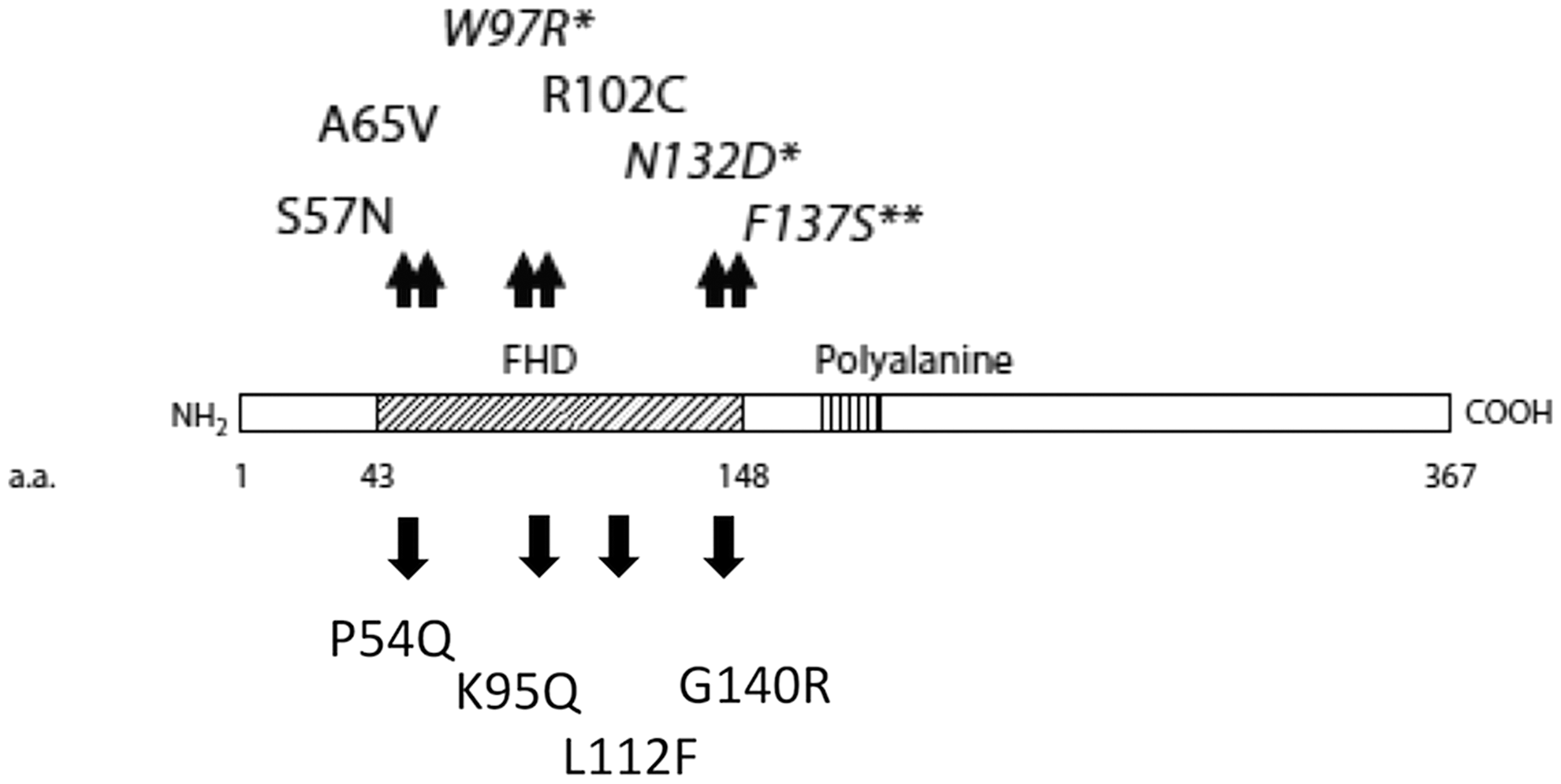
Schematic representation of the forkhead box E1 (FOXE1) gene and location of mutations identified within the forkhead domain of the protein (box). The mutations shown above the schematic are previously described inactivating germline mutations associated with Bamforth-Lazarus syndrome. The mutations shown below the schematic are novel mutations identified in papillary thyroid cancer (PTC) and multinodular goiter (MNG). Source: Adapted from Castanet and Polak, 2010 (9).
FOXE1, forkhead box E1 gene; PET, positron emission tomography.
The three PTC mutations were determined to be somatic by sequencing germline DNA isolated from patients' peripheral blood leucocytes (p.K95Q and p.L112F) or by sequencing DNA from corresponding normal thyroid tissue (p.P54Q) (data not shown). Germline DNA was not available for the patient with a MNG.
Functional characterization of the FOXE1 mutants
Transcriptional activity
In transactivation assays using the human thyroid cell line, Nthy-ori (Fig. 2B), all four mutants exhibited markedly impaired transcriptional activation on the TPO promoter, analogous to the properties of the previously characterized p.A65V mutation. Three of the four mutants were also less active on the Z16TK-LUC promoter, while the p.P54Q mutant exhibited a response equivalent to wild type FOXE1 (Fig. 2C). In HEK293 cells, the three mutants examined demonstrated markedly impaired transcriptional activation on the Z16TK-LUC promoter (Supplementary Fig. S3). Western blotting of the nuclear extracts from NThy-ori cells demonstrated that protein expression for the p.K95Q mutant was equivalent to wild type FOXE1 while the other three mutants exhibited modest reductions in protein expression in the order of 70–80% of wild type (Fig. 2A).

Molecular modeling
A theoretical structure of the FHD of FOXE1 was generated by homology modeling with FOXD3 as the template (Fig. 3B). Sequence alignment with the FHD of related proteins with known structure (Fig. 3A) indicated that the P54 and G140 residues are situated in the N-terminal and C-terminal regions of the FHD respectively, L112 localizes to the first β-sheet preceding Wing 1 while K95 is situated between the second and third α-helices. All four amino acid residues are highly conserved across species (Supplementary Fig. S4).
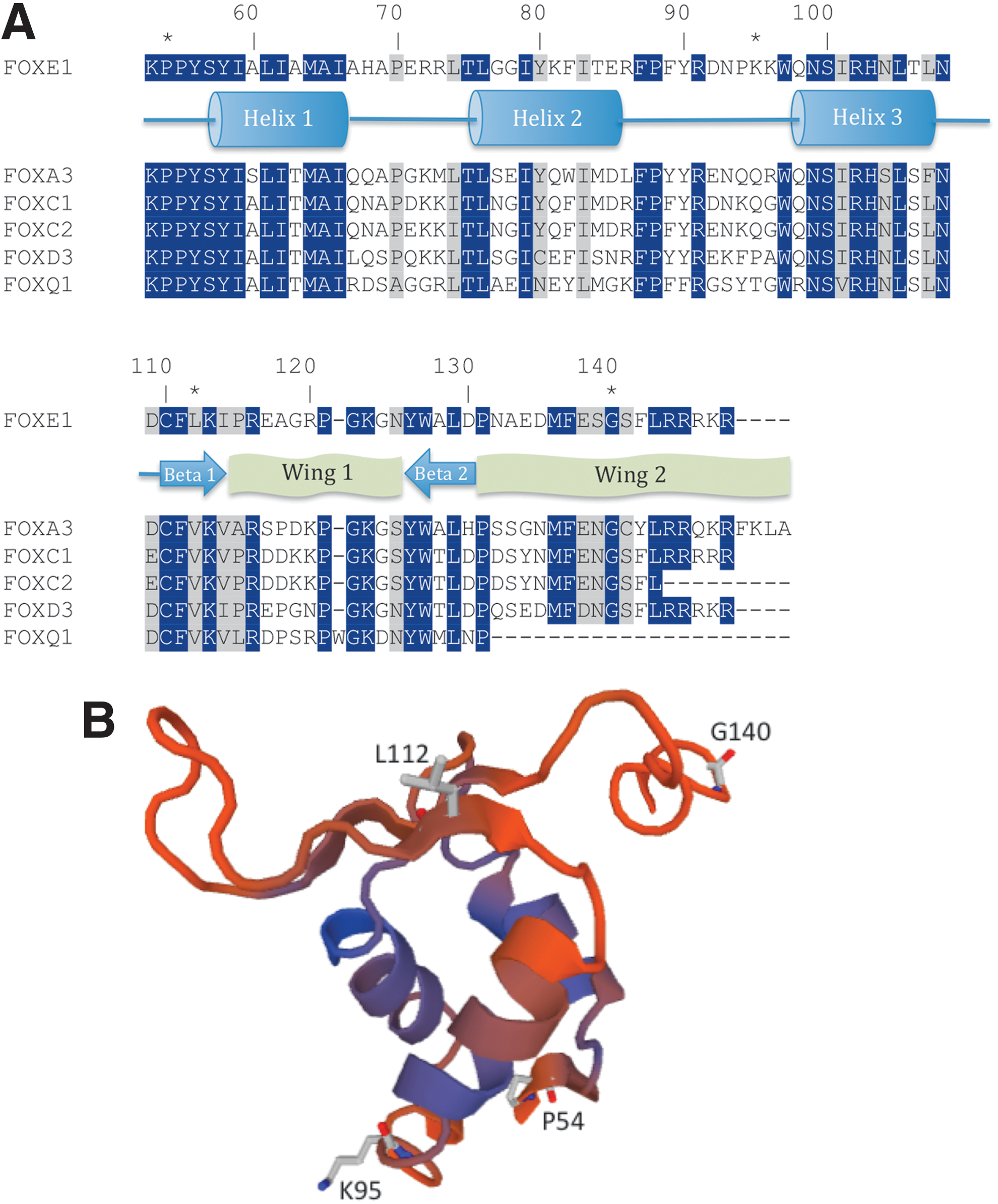
Homology modeling of FOXE1.
Discussion
In this study, we have described the functional consequences of four novel missense mutations within the FHD of FOXE1. Three of these mutations were found in PTC and were confirmed to be somatic by sequencing germline DNA or adjacent normal DNA, while one mutation was detected in MNG. All four mutations resulted in a profound impairment of transactivation function in a thyroid-specific context with the native TPO promoter. Marked impairment of transcriptional activity was also observed for the three mutants examined in HEK293 cells with the artificial Z16TK-LUC promoter. There was greater variability between the mutants in the degree of impairment on the Z16TK-LUC promoter in Nthy-ori cells, which may reflect promoter and cell context differences. Protein stability appeared to be reduced for p.P54Q, p.L112F, and p.G140R; this may reflect mutational effects on protein stability; however, the reduction in protein expression was modest and unlikely in itself to explain the dramatic impairment of transcriptional activity.
P54 and G140 are situated in the highly conserved N- and C-terminal regions of the FHD respectively. Homologous germline mutations in the corresponding nucleotides (p.P79T, p.P79L, and p.G165R) in forkhead box C1 (FOXC1) (18,19) result in Axenfeld-Rieger syndrome, which is characterized by glaucoma as well as dental and cardiac anomalies.
Consistent with our findings for p.P54Q, functional studies showed that FOXC1 p.P79T and p.P79L significantly impaired transactivation function, although DNA binding was not affected (18). P79 is situated in a segment of the FHD previously identified as a FOXC1 nuclear localization accessory signal and (incomplete) disruption in nuclear localization was observed with the mutants. The functional data was supported by molecular modeling suggesting that the proline at this position extends away from DNA making an effect on DNA binding unlikely.
Similarly, FOXC1 p.G165R did not impair DNA binding but showed significant reduction in reporter activity (36% of wild type response) (19). Molecular modeling localized G165 to the Wing 2 region of the forkhead domain with an outward facing orientation not predicted to disrupt DNA binding. Together with our data for FOXE1 p.G140R, these findings suggest an important role of the Wing 2 region in the transactivation function of forkhead proteins. The deleterious effect of mutations in this region may result from disruptions in intramolecular interactions or may be due to impaired interactions with other transcription factors.
Homology modeling suggested that the L112 residue in FOXE1 is situated in the first β-sheet, a region that is highly conserved across forkhead proteins. Loss of heterozygosity was also observed in this tumor, which may have contributed to the oncogenic potential of the p.L112F mutation. K95 is predicted to lie between the second and third helices, a region with a degree of structural variability between FOX proteins (20). However, all four mutated amino acid residues are conserved across evolution, consistent with a functionally important role within the FOXE1 protein.
Mutations in FOXE1 (different to those described in the present study) have been previously reported in individual patients with lung, stomach and squamous cell carcinoma (21,22); in some cases the mutations were confirmed to be somatic. However, the functional implications of these mutations have not been investigated and the significance of the mutations is questionable given that FOXE1 expression levels are negligible or low in these tissues.
The small number of mutations detected in our cohort suggests that mutational inactivation of FOXE1 is an uncommon event in thyroid tumors. This is reinforced by the fact that FOXE1 mutations were not reported in a recently published comprehensive genomic analysis of PTC, which included a large number of tumors (23). Of the four mutations described in the present study, one was detected in a MNG and another in a BRAF mutation-negative microPTC. The other two PTC were recurrent (BRAF V600E mutation-positive) tumors with aggressive clinical features. Review of these two cases showed evidence for tumor dedifferentiation, as both recurrences were non-iodine avid. In addition, lymph node metastases from the PTC with the p.P54Q mutation were fludeoxyglucose-avid on positron emission tomography scan, and in the case of the PTC with p.L112F mutation, histology of the recurrent tumor showed areas of dedifferentiation.
The finding of a single mutation in a benign MNG is of uncertain consequence but raises the question as to the significance of FOXE1 mutations in the pathogenesis of PTC. While the majority of nodules in a MNG are not premalignant, the condition is characterized by hyperproliferation and a significant percentage of nodules are monoclonal in origin (24). Somatic mutations have been detected in some of these nodules. Recently, germline mutations in DICER1 have been reported to predispose to familial MNG as well as a number of malignancies such as ovarian Sertoli-Leydig cell tumors (25). An association of somatic DICER1 mutations with differentiated thyroid cancer has also been reported (26). This suggests, in principle, that common genetic mechanisms could underlie a propensity to both MNG and the development of malignancy.
It is possible that somatic mutations in FOXE1 are not in themselves potent drivers of thyroid carcinogenesis but that they may, in combination with other tumorigenic events (such as a BRAF mutation), promote thyrocyte dedifferentiation, and progression of thyroid cancer. While there was evidence for dedifferentiation in the two PTC with dual BRAF and FOXE1 mutations, the small number of cases precludes any definitive conclusions. Further studies are required to investigate the possibility of a cooperative role of BRAF mutation and FOXE1 inactivation and to identify mechanisms by which such cooperation might occur. In keeping with previous reports by our group and others (3,7), we found an association between longer FOXE1 polyalanine tract variants and PTC in the present study. The notion that impairment of FOXE1 transactivation function can contribute to thyroid cancer oncogenesis is consistent with this association with FOXE116Ala being transcriptionally impaired compared with FOXE14Ala (7).
FOXE1 is part of a network of factors that help maintain thyroid differentiation via transcriptional regulation of thyroid specific genes including thyroglobulin and thyroid peroxidase (27). Recent evidence suggests that FOXE1 also regulates other genes important in thyroid function, such as dual oxidase 2 (DUOX2, which generates hydrogen peroxide required by TPO) and the sodium–iodide symporter (NIS) (28,29). Silencing of FOXE1 in a thyroid cell line was shown to result in significant reduction in NIS mRNA and protein levels (28). Loss of FOXE1 regulatory activity may therefore promote dedifferentiation of thyroid follicular cells particularly in the presence of other oncogenic drivers. It may also have therapeutic implications in that NIS and TPO are needed for radioactive iodine treatment to be effective.
In conclusion, we have identified novel somatic mutations of FOXE1 in PTC and demonstrated that these mutants impair transcriptional function. Our data reinforce the association between FOXE1 and thyroid cancer and suggest that mutational inactivation of FOXE1 may contribute to tumorigenesis in a subset of thyroid cancers.
Footnotes
Acknowledgments
The authors wish to thank Professor Krishna Chatterjee for kindly providing the FOXE1 pcDNA vector used in these studies. We also acknowledge the Victorian Cancer Biobank for assistance with tissue collection and in particular the support of Zdenka Prodanovic, Susan Hume, Yao Han, Daniela Surace, Paul Pinto Correia, and Pedro Ramos. This work was supported by the Victorian Cancer Agency through a project grant to C.G. and by the National Health and Medical Research Council (NHMRC) through a fellowship to P.J.F. (No. 1002559). M.M. is supported by Monash University though a Monash University postgraduate scholarship. MIMR-PHI Institute is supported by the Victorian government's Operational Infrastructure Program. M.B. and R.J.C.B. are supported by a NHMRC project grant (No. 1061941).
Author Disclosure Statement
No competing financial interests exist.