
Abstract
Background:
Thyrotropin (TSH) deficiency caused by TSHβ gene mutations is a rare form of congenital central hypothyroidism. Nine different TSHβ gene mutations have been reported, all with clinical manifestations. The aim was to identify the genetic cause of undetectable TSH levels in two siblings with clinical euthyroidism.
Methods:
Two brothers born to consanguineous Pakistani parents presented with undetectable serum TSH but normal iodothyronine concentrations and no clinical signs of hypothyroidism. Direct sequencing of the TSHβ gene, functional and immunological studies, protein homology modeling, and population frequency analysis were performed to characterize the cause of undetectable TSH in this family.
Results:
Direct sequencing of the TSHβ gene revealed that the two brothers were homozygous for a single nucleotide substitution (c.223A>G) resulting in the replacement of arginine 55 with glycine (R55G). This variant was found in 12 out of 5008 alleles in the 1000 Genomes project (all South Asian). Serum TSH of the two brothers was undetectable in two of five platforms, both produced by Siemens, whereas TSH levels of the heterozygous brother and mother were half compared to the other three platforms (Roche Elecsys, Abbott Architect, and Beckman Coulter DxI). The falsely low TSH concentration was caused by the monoclonal antibody not recognizing the region containing the variant amino acid. This is supported by the fact that arginine modification—following phenylglyoxal treatment—led to a significant (96%) decrease in the TSH measurement with the Siemens platforms. Predictions based on PolyPhen-2 and in silico modeling revealed no functional impairment of the variant TSH.
Conclusions:
A TSHβ variant with impaired immunoreactivity, but not bioactivity, is reported, and its biochemical impact in the homo- and heterozygous state is demonstrated. It is also shown that failure to bind to the monoclonal antibody is a direct consequence of the amino acid substitution.
Introduction
I
Clinical hypothyroidism was documented in all individuals.
The signal peptide is included in this mutation; the numbering in all other TSHβ gene mutations refers to the mature protein.
F, female; M, male; ND, not detected; Na, not available; T4, thyroxine; fT4, free thyroxine; Comp het, compound heterozygous.
A Pakistani family harboring a TSHβ variant altering the protein's immunoreactivity but not bioactivity is reported. This variant seems not to have clinical consequences but to cause misleading thyroid function tests. Its consequences in heterozygotes and the direct effect of the aa substitution on failure to bind to the monoclonal antibody are reported.
Materials and Methods
Case presentation
The proband (II-4) was a 4-year-old male, the youngest to a consanguineous Pakistani family (Fig. 1). Complaints of fatigue and low energy led to thyroid function testing. Tests revealed undetectable TSH levels (<0.004 mIU/L; Siemens Immulite 2000) with normal total thyroxine (TT4), total triiodothyronine (TT3), and free T4 index (FT4I; Fig. 1A). Thyroid imaging and pituitary function were normal.

Pedigree of the family and results of thyroid function tests and genetic analysis. (
His 10-year-old brother also had undetectable TSH with normal TT4, TT3, and FT4I and was clinically euthyroid. Both siblings had no antibodies to thyroperoxidase (TPO) and thyroglobulin (TG). Their 14-year-old brother and 17-year old sister and their mother had normal serum TSH and thyroid hormone levels. Their father declined testing (Fig. 1A).
Thyroid function tests
Blood was collected locally and shipped for analysis to the Chicago laboratory. TT4, TT3, total rT3 (TrT3), TG, and antibodies to TG and TPO were measured. FT4I was calculated from the TT4 and the resin T4 uptake ratio. TSH levels were measured with five different automated platforms (Roche Elecsys, Siemens Immulite 2000, Siemens Centaur TSH3 Ultra, Beckman Coulter DXI, and Abbott Architect).
DNA sequencing
DNA was isolated from peripheral blood leucocytes using QIAamp DNA Mini Kit (QIAGEN) followed by amplification of genomic DNA by polymerase chain reaction and direct sequencing (primers available upon request).
Testing for an interfering substance
Serum samples from the two brothers homozygous for the variant allele (II-3 and II-4), the heterozygote brother (II-2) and a normal subject were mixed at a 1:1 ratio with a pool of human sera having a TSH of 6.7 mIU/L. TSH concentration was measured with Siemens Immulite 2000 and Roche Elecsys.
TSH absorption with Immulite 2000 beads
Microparticles with biotinylated TSH antibody used in the Immulite 2000 were incubated with serum samples from a normal individual and a homozygous (II-4) and a heterozygous (II-2) individual for the variant TSHβ. The supernatant containing the TSH not bound to the particles was then assayed for TSH in the Elecsys platform.
Modification of arginine in TSH with phenylglyoxal
Phenylglyoxal (PG) was used to modify all arginine residues in human serum with normal TSH (3). TSH was eluted by addition of 0.1 M glycine pH 2.6 for 30 min at room temperature. 1 M NaHCO3 pH 9.3 was added to bring the pH to >7 followed by 0.1 M PG treatment for 1 h at 40°C.
In silico structure modeling
Three-dimensional protein homology models for the TSHβ protein, encompassing wild-type and mutant R55G, were generated using the knowledge-based method within Prime v3.2 (Schrödinger Release 2013-1) (4,5) and taking as template references the X-ray crystal structures of human FSH and human hCG. The previously described G29R mutation, known to cause CH (6,7), was also modeled and compared with the R55G. All modeling images were generated using the PyMOL Molecular Graphics System v1.5.0.4.
Results
Serum TSH levels of all family members were measured with five automated platforms (see Methods). TSH of the two brothers was undetectable using Immulite 2000 and Centaur, but was within the reference range in the other three platforms (Fig. 1B). In the Immulite 2000 and Centaur, TSH values of the two heterozygotes (I-1 and II-2) were half those obtained with the other three assays (Fig. 1B).
These findings prompted sequencing of the TSHβ gene. A single nucleotide substitution was identified (c.223A>G), resulting in the replacement of arginine 55 with glycine (GRCh37.p13: chr1:115576654A>G). Two brothers were homozygous for the alternate allele, their sister was homozygous for the normal allele, and their older brother and mother were heterozygous (Fig. 1C).
Serum samples from all male siblings were added to a human serum pool with TSH of 6.7 mIU/L to test for the presence of an interfering substance. None affected the recovery of the TSH present in the serum pool, measured by either Immulite 2000 or Elecsys.
Serum samples from normal, homozygous, and heterozygous individuals for the variant TSHβ were exposed to the Immulite 2000's microparticles coated with TSH antibody. The supernatant containing TSH not bound to the particles was assayed for TSH with Elecsys. The absorption of TSH by the monoclonal antibody in the Immulite 2000's particles was highest (82%) in the sample from the normal individual, 63% in the heterozygote, and lowest (51%) in that of the homozygote for the variant TSHβ, showing altered binding of variant TSH to the Siemens antibody.
Serum with normal TSH was treated with PG to alter arginine residues, since PG specifically modifies the guanidino group of exposed arginines in proteins (3). TSH measured by Elecsys was 4.4 mIU/L before and 4.1 mIU/L after exposure to PG, whereas with Immulite 2000 TSH decreased by 96%, from 3.9 to 0.16 mIU/L.
The mutation is tolerated/benign according to SIFT and PolyPhen-2 prediction algorithms (scores 0.53 and 0, respectively). As illustrated in 3D protein homology models (4,5) (Fig. 2), arginine 55 is pointing away from the α-subunit and the TSHR and is not expected to directly interact with either. Being distal to any of its protein partners, its replacement with glycine is not expected to alter their binding to TSHβ. However, in the deleterious G29R mutation (6,7), the normal glycine is in close proximity to the α-subunit. In the presence of the wild-type glycine, the “key-and-lock” complementarity between the two proteins is preserved. However, its change to the much larger arginine is likely to disrupt their interaction and impair the formation and/or activity of the heterodimer.
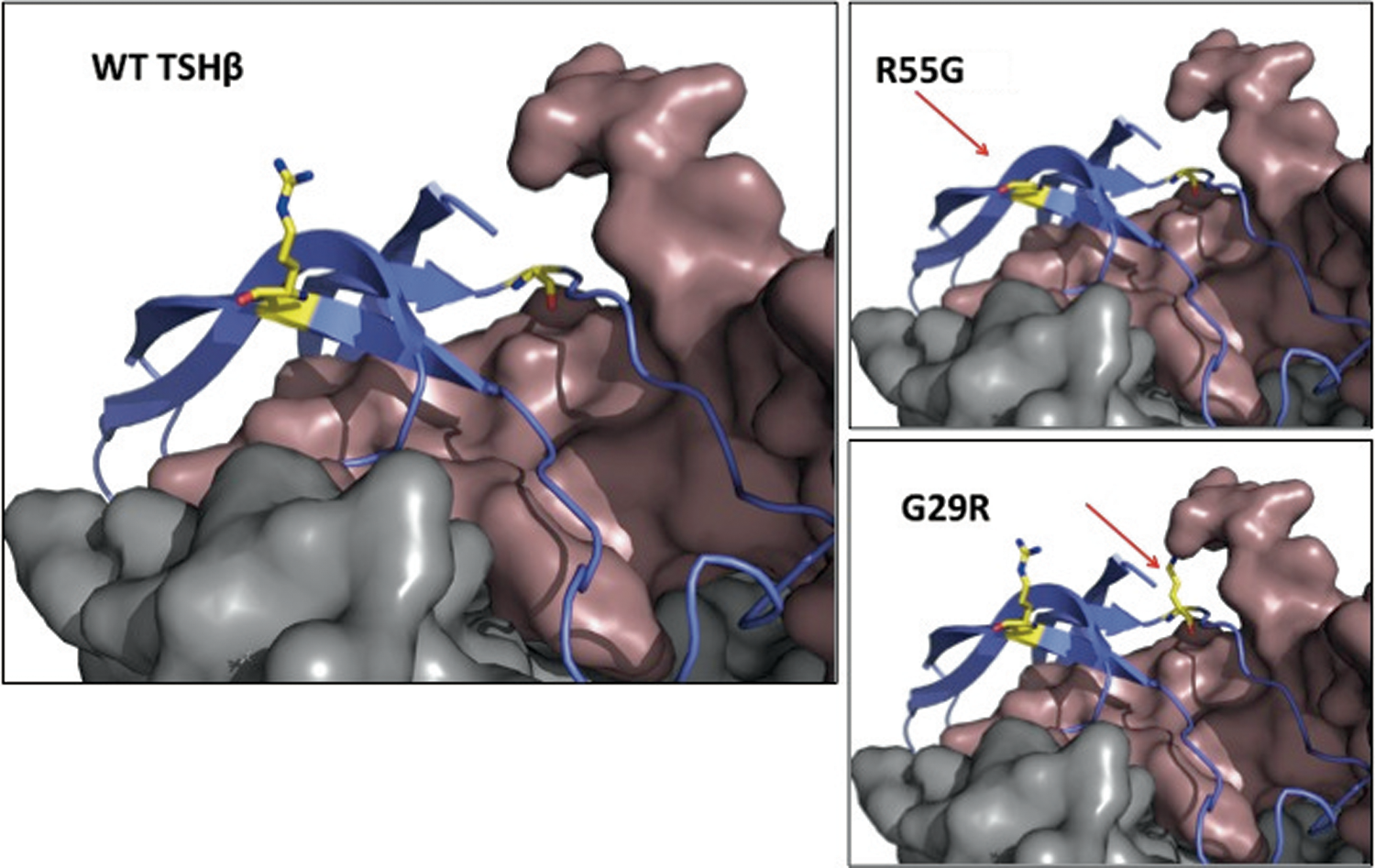
Partial representation of the 3D model of the TSH molecule (the TSH receptor in gray and the α-subunit in brown, model generated using the knowledge-based method within Prime v3.2). The normal (WT) molecule is compared to the variant R55G and the functionally impaired mutation G29R, common in Japanese (6,7). Amino acids at positions 55 and 29 are highlighted with yellow and indicated with the red arrows. All modeling images were generated using The PyMOL Molecular Graphics System v1.5.0.4. Color images available online at
Discussion
A TSHβ variant with impaired immunoreactivity but not bioactivity is reported. This variant was previously identified in South Asian individuals of Northern California during an investigation by Kaiser Permanente of incongruent thyroid function tests (8). Some patients had been inappropriately treated with antithyroid drugs. The authors demonstrated that the antibody did not recognize the mutant protein, but could not provide epitope sequence information. One of the proposed molecular mechanisms was variable glycosylation. Herein, evidence is provided that arginine loss is sufficient to alter Siemens' antibody binding to the mutant TSH. The phenotype of heterozygotes is also described, and population frequency information is provided. To the authors' knowledge, this variant is the only report of a pituitary glycoprotein with loss of immuno- but not bioactivity.
Findings supporting the normal bioactivity of the variant TSHβ include clinical euthyroidism, normal thyroid hormone levels, and normal TSH in assays other than Siemens. Two algorithms predicted that the mutation is benign, and in silico modeling showed no alteration in the interaction of TSH with the α-subunit or the TSHR (Fig. 2). This contrasts with the deleterious TSHβ mutation, G29R, prevalent in the Japanese population, in which replacement of glycine by arginine impairs the interaction with the α-subunit (6,7).
Consanguinity produced homozygosity of the variant TSH in two of four siblings and undetectable TSH in Siemens platforms. The study of the family allowed identification of two heterozygotes; although their serum TSH levels were within the reference range in the two Siemens assays, the values were half those measured by the other three analytical platforms (Fig. 1B).
Similar to the study of Drees et al., the presence of an interfering substance was ruled out, as normal serum TSH added to the sera of affected individuals was fully recovered in all assays tested. Poor TSH absorption from the serum of affected individuals by the Immulite 2000's TSH antibody-coated particles demonstrated that the variant TSH is not recognized by the Siemens antibody. Arginine modification of normally glycosylated TSH resulted in a marked TSH decrease with Immulite 2000, but not Elecsys, indicating that altered glycosylation is not involved in the loss of immunoreactivity.
The R55G variant was observed in a heterozygous state in the 1000 Genomes project (12/5008 alleles, minor allele frequency [MAF] 0.0024) and the ClinSeq Agilent project (1/1324 alleles, MAF 0.0007). In the 1000 Genomes database, it was found only in South Asian individuals from Bangladesh, India, Sri Lanka, and Pakistan with a subpopulation MAF of 0.012, fivefold more frequent compared with the general population. This is relevant, since the family is of Pakistani origin. Considering a distribution following the Hardy Weinberg equilibrium, the calculated MAF in the study of Drees et al. (8) is 0.0035, similar to those above.
To date, nine different TSHβ gene mutations have been reported. All individuals with biallelic mutations had clinically overt central hypothyroidism. A detailed review of all previously reported TSHβ gene mutations is presented in Table 1. These mutations include three missense variants and six producing a premature stop codon (two as a result of frame-shift, two due to single nucleotide polymorphisms, and two splice-junction mutations) (6,7,9 –31). Interestingly, in a report of the Q49X mutation, in which the product lacks 60% of the C-terminal tail of the mature protein, TSH could, in part, be measured by several immunoassays; the levels ranged from undetectable (when third-generation immunoradiometric and immunofluorometric assays where used) to normal (with second-generation assays and a third-generation electrochemiluminescence immunoassay) (31). Despite the lack of bioactivity, the mutant Q49X TSHβ protein could apparently form a heterodimer with the α-subunit conferring some degree of immunoreactivity and thus allowing detection of the mutant protein by some immunoassays. Similarly, there are few other reports that, depending on the immunoassay, detected normal or even high serum TSH concentrations in individuals with central hypothyroidism harboring TSHβ gene mutations (Table 1). In the clinical setting, these normal TSH measurements could be misleading and delay diagnosis of congenital central hypothyroidism.
Novel data in this study include TSH measurement in heterozygotes for the R55G variant, being half with the Siemens platform compared to the other assays. The significant decrease in TSH (with the Siemens assay) after PG treatment makes a glycosylation defect an unlikely scenario and points toward the lack of recognition of arginine 55 in the TSHβ epitope by the assay antibody as the cause of spuriously low TSH. Protein homology modeling further supports the normal structure of the TSHβ variant. Lastly, population frequency data are presented from three different projects with similar MAFs and a higher frequency in the South Asian population.
In conclusion, a TSHβ variant with impaired immunoreactivity but normal bioactivity was identified in a Pakistani family. Using methods that do not recognize the variant TSH could lead to an erroneous diagnosis and potentially to inappropriate treatment (8).
Footnotes
Acknowledgments
This work was supported in part by Grant R37DK15070 from the National Institutes of Health and the Seymour J. Abrams fund for thyroid research. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Diabetes and Digestive and Kidney Diseases or the National Institutes of Health.
Author Disclosure Statement
No competing financial interests exist.