
Abstract
Background:
It can be useful to know the transgene insertion site in transgenic mice for a variety of reasons, but determining the insertion site generally is a time consuming, expensive, and laborious task.
Methods:
A simple method is presented to determine transgene insertion sites that combines the enrichment of a sequencing library by polymerase chain reaction (PCR) for sequences containing the transgene, followed by next-generation sequencing of the enriched library. This method was applied to determine the site of integration of the thyroid peroxidase promoter–Cre recombinase mouse transgene that is commonly used to create thyroid-specific gene deletions.
Results:
The insertion site was found to be between bp 12,372,316 and 12,372,324 on mouse chromosome 9, with the nearest characterized genes being Cntn5 and Jrkl, ∼1.5 and 0.9 Mbp from the transgene, respectively. One advantage of knowing a transgene insertion site is that it facilitates distinguishing hemizygous from homozygous transgenic mice. Although this can be accomplished by real-time quantitative PCR, the expected Ct difference is only one cycle, which is challenging to assess accurately. Therefore, the transgene insertion site information was used to develop a 3-primer qualitative PCR assay that readily distinguishes wild type, hemizygous, and homozygous TPO-Cre mice based upon size differences of the wild type and transgenic allele PCR products.
Conclusions:
Identification of the genomic insertion site of the thyroid peroxidase promoter–Cre mouse transgene should facilitate the use of these mice in studies of thyroid biology.
Introduction
T
The authors' laboratory studies a form of thyroid cancer caused by a gene fusion between PAX8 and PPARG, which results in production of an oncogenic PAX8–PPARG fusion protein, PPFP (1). The authors have created a mouse model of this disease in which expression of a transgenic PPFP cDNA and deletion of endogenous Pten (thereby activating Akt) are dependent on expression of Cre recombinase (2). Mice that contain the Cre-dependent PPFP transgene and that have homozygous floxed Pten alleles are bred to mice which are homozygous both for floxed Pten and for a transgene in which the thyroid-specific thyroid peroxidase (TPO) promoter drives expression of Cre (3). Mice that are homozygous for TPO-Cre are preferred over hemizygous mice because it allows the breeding scheme to be much more efficient. However, it is difficult to distinguish homozygous from hemizygous transgenic mice unambiguously by quantitative real-time PCR, since the expected difference in Ct values is only one cycle. Identification of the transgene insertion site would facilitate the development of a qualitative PCR assay to distinguish homozygotes from hemizygotes.
For these reasons, a combination of PCR enrichment of a sequencing library with next-generation sequencing of the PCR products was used to circumvent these limitations and to identify the genomic insertion site of the TPO-Cre transgene. This approach should facilitate the determination of any transgene insertion site.
Materials and Methods
Transgenic TPO-Cre mice
All animal husbandry procedures were approved by the Institutional Animal Care and Use Committee of the University of Michigan. Transgenic mice in which the TPO promoter drives the expression of Cre recombinase (TPO-Cre) have been described (3). This transgene contains the human TPO promoter, Cre recombinase. and mouse Mt1 polyadenylation sequences, flanked by dual chicken HS4 insulators.
Generation of DNA sequencing library and next-generation sequencing
Liver DNA from a hemizygous or homozygous TPO-Cre mouse was prepared using a Promega Wizard Genomic DNA Purification Kit. A DNA sequencing library was prepared in the University of Michigan DNA Sequencing Core from that genomic DNA. Briefly, the DNA was sheared using the Covaris S2 system, and fragments of approximately 550 bp were isolated by agarose gel electrophoresis, excision, and purification. The purified DNA was converted to a sequencer-ready library using the NEB-Next DNA Sample Prep Kit (New England Biolabs), according to the manufacturer's recommended protocols. One μL of this library was amplified by 35 cycles of PCR using Promega GoTaq G2 Polymerase and cycling conditions of 95C × 3 min; then 35 cycles of 95C × 30 sec, 60C × 30 sec, and 72C × 1 min, and then 72C × 5 min. One PCR primer is denoted LP2 and is identical to Illumina's “TruSeq PCR Primer 2” (see Table 1), and the other primer is denoted TG1 and is directed at sequences within the 5′ end of the transgene. Because the LP2 primer is only present on one end of each library fragment, it is unable to initiate exponential amplification by itself. Only when the gene-specific (TG1, in this case) primer also is present will exponential amplification be possible. All primer sequences are provided in Table 1, and a schematic representation of the PCR strategies is shown in Figure 1.
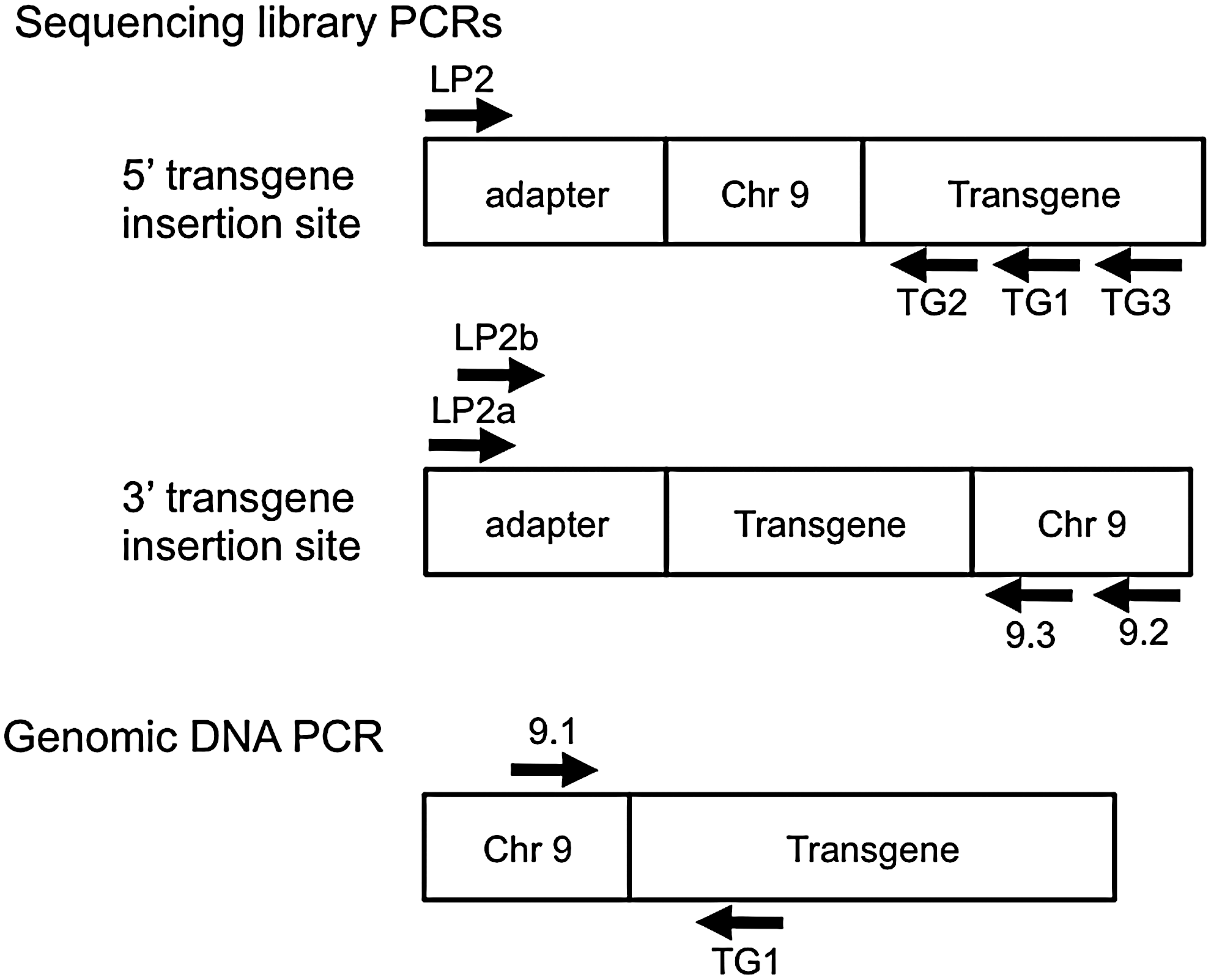
Schematic representation of polymerase chain reaction (PCR) primers used in determination of the transgene insertion site. Hypothetical sequencing library molecules are shown that contain the library adapter and a genomic fragment that includes either the 5′ or 3′ junction of the transgene with the mouse genomic DNA. The positions of PCR primers are shown. A similar schematic is shown to illustrate primers used to PCR the 5′ insertion site from genomic DNA. The schematics are not drawn to scale.
The product of this PCR was diluted 1000-fold in water, and 1 μL was subjected to a second round of PCR that utilized LP2 with a new transgene-specific primer, TG2, that was nested relative to TG1 (i.e., closer to the 5′ end of the transgene). As a specificity control, the transgene-specific primer was replaced by primer TG3, located farther away from the 5′ end of the transgene and therefore expected to yield no product. These reaction products were subjected to agarose gel electrophoresis to confirm successful amplification of DNA only from the appropriately nested reaction, and that material was then subjected to next generation sequencing using a Pacific Biosciences RS II sequencing system. Briefly, libraries were created using Pacific Biosciences recommended protocols for small fragments, and sequenced on the RS II according to the manufacturer's recommended protocols for diffusion loading, using the P4/C2 chemistry with circular consensus data analysis. The resulting FASTA files were analyzed manually, via a simple text search to identify sequence reads containing the TG2 primer sequence. Sequences found adjacent to TG2 were then manually submitted to NCBI BLAST to identify any putative mouse chromosomal sequence.
As described in the Results, this yielded a tentative identification of the integration site of the 5′ end of the transgene in mouse chromosome 9. A PCR primer pair was then created to confirm this, with one primer being in the mouse genome (primer 9.1) and the other in the transgene (TG1); the product was subjected to conventional Sanger sequencing. Similar approaches were used to identify the 3′ integration site, as presented in the Results.
Results
Identification of the TPO-Cre transgene genomic insertion site
Only a tiny fraction of the DNA molecules in a sequencing library will contain the junction between the host chromosome and the transgene. To enrich for these molecules, nested PCR was utilized. In the first reaction, one primer (LP2) was a subsequence of the adapter used in construction of the library, and the other primer was directed at transgene sequences near the 5′ end of the transgene (primer TG1; Fig. 1). Amplification of the sequencing library with these primers is expected to have only modest specificity, since primer LP2 will anneal to all DNA molecules in the library. Therefore, the products of this PCR were subjected to a second round of PCR that used LP2 with transgene-specific primer TG2 that was nested relative to TG1 (i.e., closer to the 5′ end of the transgene). As shown in Figure 2, this resulted in a smear of products, which is the expected result, since the distance between TG2 and the 5′ end of each library molecule will be variable. As a specificity control, primer TG2 was replaced with TG3, which is located external to TG1, and hence should not be able to amplify true transgene products from the first round of PCR. This yielded very little product, as expected (Fig. 2).
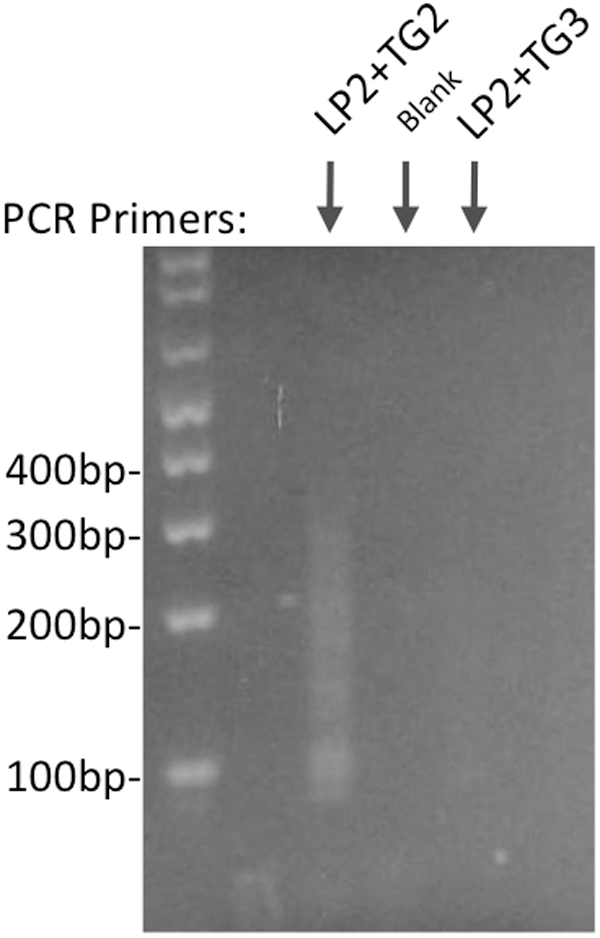
Agarose gel electrophoresis of PCR products obtained from nested PCR of sequencing library DNA. A first round of PCR of the sequencing library DNA (not shown) was performed using as one primer sequences from the oligonucleotide adapter used for library construction (LP2), and the second primer was directed at transgene sequences near the 5′ end of the transgene (TG1). The PCR product was diluted 1000-fold, and 1 μL was used in a second round of PCR with the same library adapter primer (LP2) plus a transgene-specific primer that is nested relative to the round 1 PCR primer (TG2). This PCR yielded a smear of DNA ∼100–300 bp. As a specificity control, TG2 was replaced by TG3, which is directed at transgene sequences external to the first round PCR product.
The products of the nested PCR with primers LP2 and TG2 were subjected to sequencing using a Pacific Biosciences RS II sequencing system, and sequencing reads were found (by manual search) that contained both transgene-specific primer and mouse chromosomal sequences. This indicated that the transgene is on chromosome 9, oriented on the reverse strand, with chromosome 9 bp 12,372,324 adjacent to the 5′ end of the transgene (Fig. 3). To confirm this insertion site, genomic DNA from a TPO-Cre mouse was PCR amplified using one primer from chromosome 9 (primer 9.1) and one primer from the transgene (TG1). The PCR product was subjected to Sanger sequencing, confirming the 5′ transgene integration site (Fig. 3).

Junction of chromosome 9 and the TPO-Cre transgene at the sequence level. Schematic shows the sequence of chromosome 9 top strand whereas the transgene is inserted in the opposite orientation.
The transgene 3′ insertion site was then identified in the following manner. The sequencing library was subjected to PCR using a primer directed at chromosome 9 downstream from the transgene 5′ integration site (primer 9.2; Fig. 1) with a shortened version of LP2 denoted LP2a (shortened to create a more optimal primer pair). Similar to the approach to identify the 5′ insertion site, this PCR product was subjected to a second round of PCR using a chromosome 9 primer (9.3) that is nested relative to primer 9.2 with another shortened version of LP2, denoted LP2b. The PCR product was subjected to Sanger sequencing using primer 9.3, which identified the 3′ transgene insertion site (Fig. 3). Note that transgene integration resulted in deletion of chromosome 9 bp 12,372,317–12,372,323.
Development of a qualitative PCR assay to distinguish hemizygous from homozygous TPO-Cre mice
To maximize efficiency, the authors' laboratory uses homozygous TPO-Cre mice in breeding schemes, as noted above. However, it is difficult to distinguish homozygous from hemizygous TPO-Cre mice by quantitative real-time PCR, since the expected difference in Ct values is only one cycle. The genomic insertion site information was therefore used to develop a qualitative PCR assay that can distinguish wild type, hemizygous, and homozygous TPO-Cre mice. This PCR utilizes chromosome 9 primers 9.1 and 9.4, which flank the transgene, and one transgene primer (TG1; Fig. 4A). This PCR yields a 379 bp product for the wild type allele and a 332 bp product for the transgenic allele. Agarose gel electrophoresis of the PCR products reliably distinguishes wild type, hemizygous, and homozygous TPO-Cre mice (Fig. 4B).
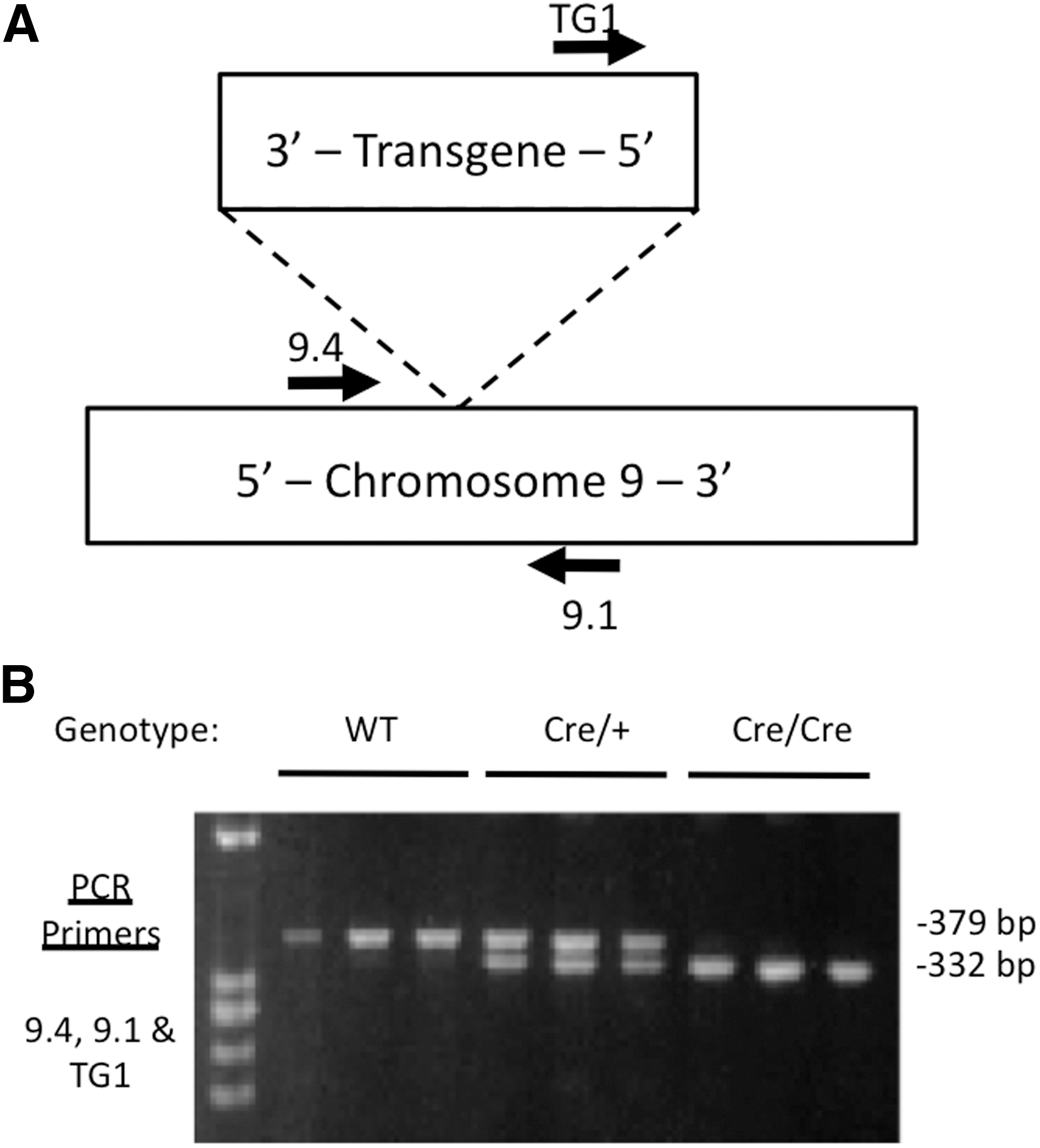
Qualitative PCR genotyping of wild type, hemizygous, and homozygous TPO-Cre mouse DNA. (
Discussion
A simple, efficient method has been developed to determine the insertion site of transgenes in transgenic mice, and this has been applied to identify the insertion site of the TPO-Cre transgene that is widely used to achieve thyroid-specific expression of Cre. The insertion site was found to be between bp 12,372,316 and 12,372,324 on mouse chromosome 9, with the nearest characterized genes being Cntn5 and Jrkl, ∼1.5 and 0.9 Mbp from the transgene. Given these large distances, it is very unlikely that the transgene will affect Cntn5 or Jrkl gene expression. Cntn5 encodes a neuronal membrane protein that functions as a cell adhesion molecule, and the function of the protein encoded by Jrkl is not known. There are no known associations between these proteins and thyroid biology. An advantage of this method is that PCR enrichment of the sequencing library for transgene-containing sequences allows for relatively shallow sequencing, thus decreasing the cost and simplifying the data analysis. There are multiple advantages to knowing a transgene insertion site. For example, the transgene could potentially disrupt the expression of nearby genes, or nearby sequences could influence the expression of the transgene. In the present case, the authors wished to develop a robust, qualitative assay to distinguish hemizygous from homozygous TPO-Cre mice, and knowledge of the transgene insertion site made it easy to establish such an assay that can be run using conventional PCR.
A potential weakness of this method is that the 5′ and 3′ ends of the transgene are not always what one would predict based upon the construct used to create the mice. The reason for this is that the transgenic DNA usually concatemerizes such that multiple head to tail copies are present within the insertion site. The first or last copy of the transgene may not be complete, and in fact, for TPO-Cre, it was found that the 3′ most copy of the transgenic insert is partial and terminates in the middle of the TPO promoter. This is a potential limitation because to PCR-enrich the sequencing library one must use nested primers that are near either the 5′ or 3′ end of the inserted transgene. However, this is a minor issue because only one of the two ends of the transgene has to be enriched, and several different primers can be tried. In addition, any PCR-based strategy, including the present one, may be difficult to apply if the transgene is flanked by sequences with repetitive elements or has a high GC content resulting in poor PCR performance.
Classical methods to determine transgene insertion sites have utilized chromosome walking. A number of PCR-based methods are available for chromosome walking, such as inverse PCR (4), ligation-mediated PCR (5,6), and specific-primer PCR (7,8), which identify transgene flanking sequences. However, these methods are inefficient, can generate nonspecific amplification products, and have other deficiences that make them difficult to apply (9). Other researchers have used a combination of microarray hybrid capture and next-generation sequencing to identify transgene insertion sites (10). This method, however, requires designing a custom microarray for targeted sequence capture and enrichment. Shallow coverage whole genome sequencing has also been carried out in combination with searching for tandem duplications to help identify the transgenic insert, although this method would not be feasible for transgenic inserts without tandem repeats (11). The present method of combining PCR with next-generation sequencing helps to enrich the transgenic insertion sites and thus is cost-effective and efficient. The predicted genomic integration sites allow for designing a fast and convenient PCR method to determine and distinguish the zygosity status of the transgenic mice studied.
Footnotes
Acknowledgments
We thank Dr. Shioko Kimura for providing the TPO-Cre mice. This work was supported by NIH grant R01CA166033.
Author Disclosure Statement
No competing financial interests exist.