
Abstract
Background:
The firefly luciferase reporter protein is a crucial tool for studies targeting a broad range of biological questions. Importantly, luciferase assays are also widely used to explore mechanisms underlying thyroid hormone dependent regulation of gene expression. However, it was demonstrated that the firefly luciferase reporter is subject to triiodothyronine (T3)-evoked, promoter independent downregulation that is mediated by the thyroid hormone receptor. Since this effect can interfere with readout accuracy, the study aimed to find luciferase reporters that are not susceptible to this phenomenon.
Methods:
Luciferase reporter constructs were generated under the control of a minimal thymidine kinase (TK) promoter and transiently transfected into JEG-3 cells to test their activity upon T3 treatment.
Results:
Activity of the TK-(dCpG)Luc encoding a synthetic (dCpG)Luciferase and TK-NanoLuc expressing the NanoLuc reporter was not significantly changed by T3 treatment while the firefly luciferase control was suppressed by ∼2.6-fold. T3 also downregulated the activity of Renilla luciferase by ∼30%.
Conclusions:
Novel types of luciferase reporters, especially the synthetic (dCpG)Luciferase, can be more accurate to study T3-regulated gene expression than the classical firefly luciferase reporter. Renilla luciferase, a popular transfection control of dual luciferase assays, should be used with caution in conditions with T3 treatment.
Introduction
T
Materials and Methods
Plasmids
The pTRE-TK-Luc, which contains the thymidine kinase (TK) minimal promoter of the herpes simplex virus, was kindly provided by Dr. A.M. Zavacki (Boston, MA). The TK-Luc construct was generated by removing the TRE triplet of pTRE-TK-Luc by digestion with BamHI and BglII followed by religation and confirmation of the final construct by direct sequencing.
TK-(dCpG)Luc was prepared using the TK-Luc plasmid backbone as follows. The pMOD Luc-ShS v02 plasmid (InvivoGen, San Diego, CA) was used as a template to amplify the (dCpG)Luc coding region with Vent PCR (oligos: sense, catgcc ATG GAG GAT GCC AAG AAT ATT AAG AA; antisense, gga att cTT ATT TGC CAC CCT TCT TGG CCT TGA TCA). The amplicon was cut with NcoI and inserted into the NcoI– and the blunted EcoNI sites of TK-Luc. The construct was confirmed by sequencing.
TK-Renilla-Luc was generated by truncating the 760 bp-long TK promoter of pRL-TK (Promega, Madison, WI) using BglII and EcoRI digestion followed by blunting with Klenow polymerase and subsequent religation. This resulted in a minimal TK promoter between EcoRI and HindIII that is 31 bp shorter than the 128 bp-long minimal TK promoter of TK-Luc. 3′ to the TK promoter, this construct also contains a 136 bp chimeric intron originating from pRL-TK. The construct was confirmed by restriction mapping.
TK-NanoLuc was prepared by isolating the TRE lacking the minimal TK promoter from the pTRE-TK-Luc through digestion with BglII and HindIII, subsequent cloning of the released fragment into the respective sites of the pNL1.1 vector (Promega), and confirmation of the final construct by sequencing.
A mouse TRα (mTRα) expression construct was generated using the TRαCDM plasmid (7) (kindly provided by Dr. A.M. Zavacki, Boston, MA) as a template to amplify mTRα coding region with Vent PCR (oligos sense: gga att cca ttA TGG AAC AGA AGC CAA GCA AGG T; antisense: ata aga atg cgg ccg cTT AGA CTT CCT GAT CCT CAA AGA). The amplicon was cut with EcoRI and NotI and inserted into these sites of a pCI-Neo vector (Promega) and final confirmation by sequencing.
The secreted embryonic alkaline phosphatase (SEAP) encoding pSEAP2-Promoter plasmid (Clontech, Mountain View, CA) was used for transfection control.
Cell culture and transfection
JEG-3 human choriocarcinoma cells (kindly provided by Dr. J. Szekeres, Pécs, Hungary) were cultured in 24-well plates in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS). When ∼70% confluency was reached, cells were transfected with 800 ng DNA/well (including 200 ng Luciferase reporter, 100 ng mouse TRα, 10 ng pSEAP2, and 490 ng pUC as inert DNA) using Lipofectamine® 2000 (Life Technologies/Thermo, Carlsbad, CA). After about six hours, the transfection media was replaced with DMEM containing 10% hormone-free FBS, prepared as previously described (8), and incubated for 40 hours. The media were replaced with DMEM with 10% hormone-free FBS containing either 50 nM 3,5,3′-triiodothyronine (+T3) or NaOH vehicle (−T3). After 24 hours, the culture media were collected for SEAP measurement. The cells were washed with PBS and harvested in 100 μl passive lysis buffer (Promega).
Assays
Luciferase activity was measured from 20 μl cell lysate with the Dual-luciferase Reporter Assay System (firefly luciferase for pTRE-TK-Luc, TK-Luc, TK-(dCpG)Luc; Renilla luciferase for TK-Renilla-Luc) as previously described (9). Activity of the TK-NanoLuc was determined from 20 μl cell lysate with the Nano-Glo Luciferase Assay System (Promega), according to the manufacturer's instructions. Luciferase activity of each transfected well was assayed separately. Measurements with the Dual-luciferase Reporter Assay System were performed in duplicates that were averaged to express luciferase activity of each well. Duplicates were closely agreeing with an average difference of 7 ± 1.62% and 5.9 ± 1.2% (mean ± standard error of the mean [SEM]; n = 14) for −T3 and +T3 groups of pTRE-TK-Luc, respectively. All measurements were performed with a Luminoskan Ascent Luminometer (Thermo, Waltham, MA).
SEAP activity was determined from 25 μl media with Nova Bright™ SEAP Enzyme Reporter Gene Chemiluminescent Detection system 2.0 (Invitrogen/Thermo) as previously described (8). SEAP was used for normalization by calculating Firefly luciferase (Luc)/SEAP light unit or Renilla luciferase (Renilla)/SEAP light unit ratios for each well. Experiments were performed at least eight times, and data are presented as mean ± SEM.
Statistics
Statistical analysis was performed with an unpaired two-sample t-test using a 95% level of confidence.
Results
Different types of luciferase reporter mammalian expression constructs were generated governed by a minimal TK promoter (Fig. 1). First, the conditions were tested in JEG-3 cells with pTRE-TK-Luc and the TK-Luc firefly luciferase in the presence of co-transfected mTRα. T3 treatment (50 nm) induced luciferase activity of the pTRE-TK-Luc around threefold, while the same treatment suppressed luciferase activity of the TK-Luc encoded reporter ∼2.6-fold (Fig. 2). In contrast, T3 treatment did not affect the level of the SEAP internal control (SEAP level in pTRE-TK-Luc wells −T3: 0.97 ± 0.078 vs. +T3: 0.79 ± 0.084; mean ± SEM; n = 14, p = 0.13; SEAP level in TK-Luc wells −T3: 0.86 ± 0.105 vs. +T3: 0.76 ± 0.057; mean ± SEM; n = 8, p = 0.4).
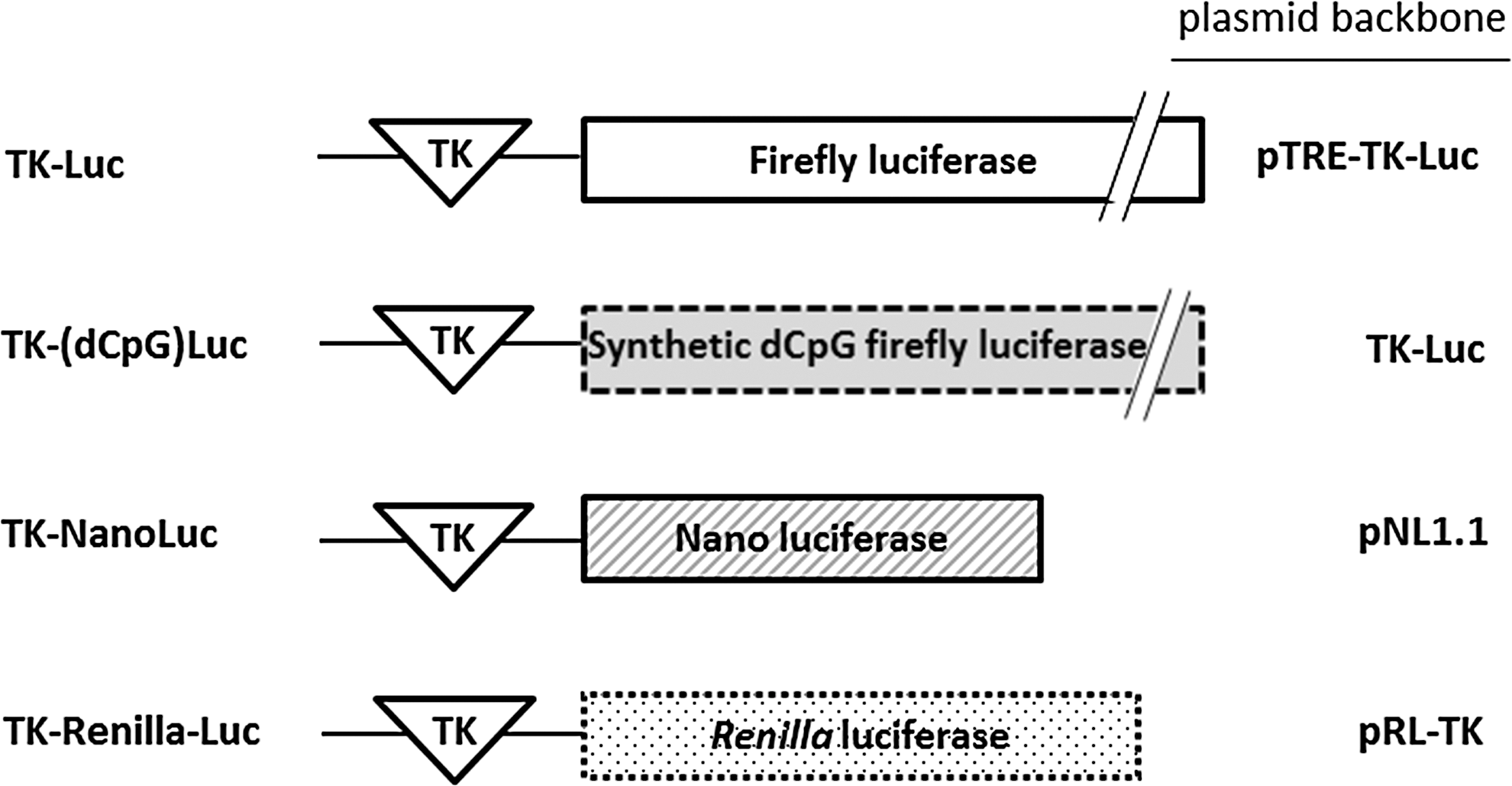
Schematic depiction of mammalian expression constructs encoding different types of luciferase reporters driven by a minimal thymidine kinase (TK) promoter. The harboring plasmid backbone is indicated. The firefly luciferase cDNA of TK-Luc was replaced with the coding region of dCpG firefly luciferase. Thus, TK-Luc and TK-(dCpG)Luc are identical except for the regions encoding luciferase.
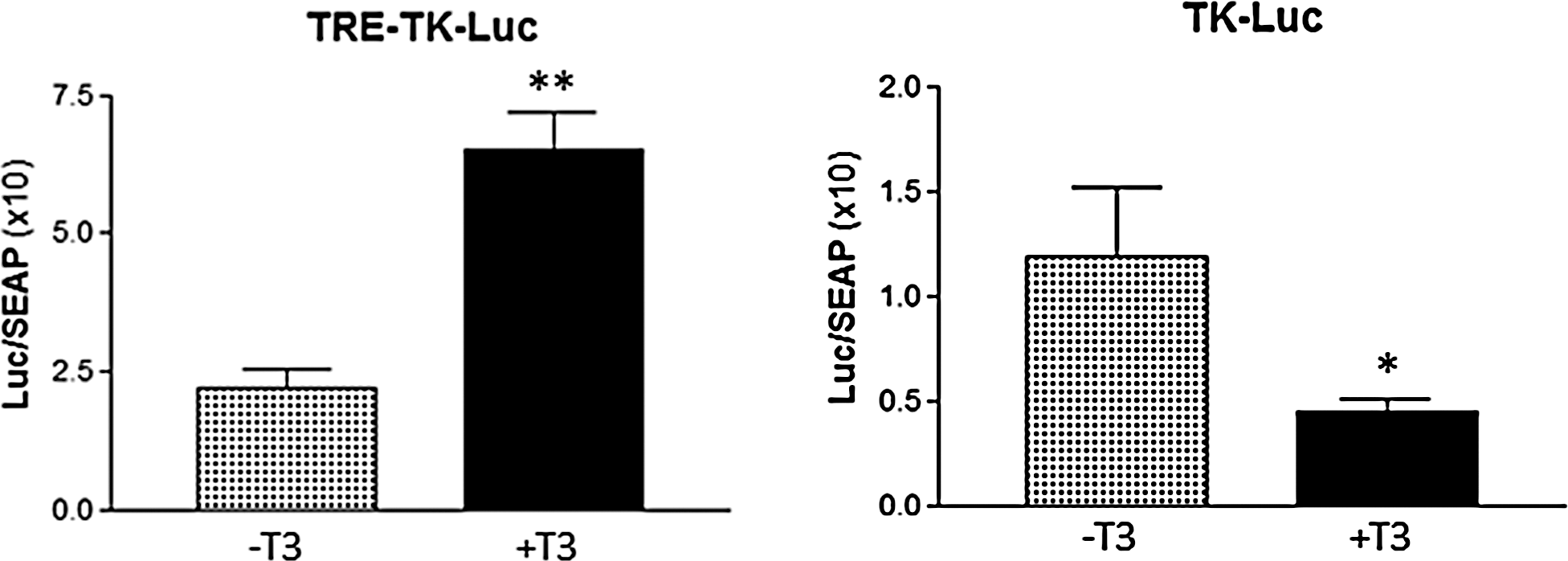
Response of pTRE-TK-Luc and TK-Luc control firefly luciferase reporters to 50 nM T3 in JEG-3 cells. Reporters were co-transfected with mouse TRα as described in the Materials and Methods. T3 induced the luciferase activity of the pTRE-TK-Luc around threefold (n = 14; p < 0.001). The activity of TK-Luc was suppressed ∼2.6-fold (n = 8; p < 0.05). Results are expressed as firefly luciferase (Luc)/SEAP light unit ratios as mean ± standard error of the mean (SEM).
To identify luciferase reporters lacking unwanted T3 regulation, the same approach was used to test how other constructs encoding different types of luciferase reporters respond to T3 in the absence of a TRE in the canonical position 5′ to the promoter sequence. Importantly, the TK-(dCpG)Luc encoding a synthetic firefly luciferase was not affected by 50 nM T3. Upon T3 treatment, the luciferase activity of the TK-NanoLuc showed a slight tendency of decreased activity, but it did not reach significance (Fig. 3). In contrast, T3 treatment significantly decreased the activity of cultures expressing the TK-Renilla-Luc by ∼30% (Fig. 3).
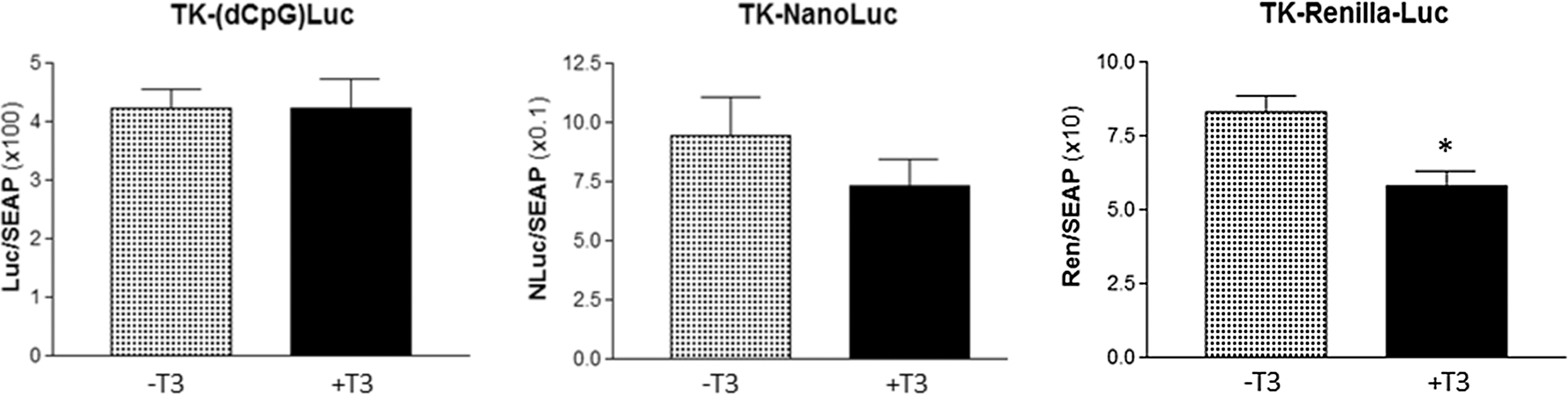
Response of TK-(dCpG)Luc, TK-NanoLuc and TK-Renilla Luc luciferase reporters to 50 nM T3 in JEG-3 cells. Reporters were co-transfected with mouse TRα as described in the Materials and Methods. T3 did not significantly affect luciferase activity encoded either by the TK-(dCpG)Luc (n = 28; p = 1) or TK-NanoLuc (n = 12; p = 0.27). T3 significantly reduced the activity of the TK-Renilla-Luc reporter (n = 11; p < 0.005). Results are expressed as firefly, Nano, or Renilla luciferase (Luc, NLuc, or Ren, respectively)/SEAP light unit ratios as mean ± SEM.
Discussion
Luciferase reporters are widely used to dissect mechanisms underlying T3-mediated regulation of gene expression. However, the limitation of this approach has been shown previously by demonstrating both a TRα and TRβ-dependent but promoter-independent negative response of firefly luciferase possibly through an unidentified negative TRE embedded in the coding region of the reporter (3,4). Negative TREs are heterogeneous and poorly defined. Thus, computer-assisted sequence analysis cannot be efficiently used for their detection.
Since different luciferase reporters became available, this study aimed to test their susceptibility to T3 treatment. The Herpes Simplex minimal TK promoter was used, since it was shown that is unaffected by T3 (3). The reporters were transiently expressed in JEG-3 cells because this cell line was found to be a sensitive model for T3-mediated downregulation of firefly luciferase (4). In agreement with earlier findings (3,4), T3-mediated downregulation of firefly luciferase under the applied conditions in the presence of co-transfected TRα was also observed. Using this system, it was demonstrated that the (dCpG) luciferase is not subjected to T3-induced downregulation. This synthetic reporter gene is a codon-optimized firefly luciferase that encodes a protein with an identical amino acid sequence with firefly luciferase except that it lacks its carboxy-terminal three amino acids. However, in order to make it more suitable for mammalian expression, its coding sequence was heavily mutated to optimize codon usage, and all the 95 of 2′-deoxyribo-cytidine-phosphateguanosine (CpG) dinucleotides present in the original sequence of firefly luciferase have been removed. Due to these numerous mutations, the resulting coding region is only 77% identical with the sequence encoding wild type firefly luciferase (Fig. 4). It is assumed that the negative TRE embedded in the coding region of wild type firefly luciferase has been disrupted through the insertion of these mutations.

Alignment of the 5′ 360 bp-long fragment of the coding region of firefly luciferase to the (dCpG) Luciferase. The full coding regions are only 77% identical but encode the same amino acid sequence.
It was also found that NanoLuc, a novel luciferase isolated from the deep-sea shrimp Oplophorus gracilirostris (10), is not significantly affected by T3. The detected minimal change in its activity is in the range where the applied assay reaches its limits of resolution. This NanoLuc reporter does not share any sequence similarity with firefly luciferase, and the present findings indicate that it also lacks a potent negative TRE. Finally, Renilla Luciferase, which originates from the sea pansy Renilla reniformis (11), was also tested. Renilla expression was significantly suppressed by T3, although this was less pronounced than the observed decrease of the wild type firefly luciferase. Since Renilla luciferase is a popular internal transfection control in dual luciferase assays, its regulation by T3 should be taken into account.
In summary, the present findings indicate that different types of luciferase reporters show different susceptibility to T3 induced downregulation. Careful selection of reporter constructs and novel luciferase reporters allow a more reliable readout in studies targeting T3-mediated regulation of gene expression.
Footnotes
Acknowledgments
The technical help of A Juhász and P Mohácsik is gratefully acknowledged. JEG-3 cells were kindly provided by Dr. J. Szekeres (Pécs, Hungary), and the pTRE-TK-Luc and TRαCDM plasmids by Dr. A.M. Zavacki (Boston, MA). This work was supported by the Hungarian National Brain Research Program and Hungarian Scientific Research Fund OTKA K109415.
Author Disclosure Statement
The authors do not have anything to disclose.