
Abstract
Background:
Differentiated thyroid carcinomas (DTC) are associated with a good prognosis and a high survival rate. However, tumor recurrence occurs in approximately 20–30% of DTC patients, reinforcing the importance of identifying new molecular targets for cancer management. It has been shown that the 5′-AMP-activated protein kinase (AMPK) is over-activated in papillary thyroid cancer (PTC). This study aimed to investigate the effects of 5-aminoimidazole-4-carboxamide-ribonucleoside (AICAR), an AMPK activator, on various aspects of thyroid cancer cell behavior, including cell survival, apoptosis, migration, invasion, and epithelial-to-mesenchymal transition (EMT), in the human thyroid cancer cell lines BCPAP and TPC-1.
Methods:
BCPAP and TPC-1 cells were cultivated in Dulbecco's modified Eagle's medium, and the non-tumor-derived cell line Nthy-ORI was grown in RPMI. Cells were treated or not with AICAR for different periods of time. The cell growth rate, cell cycle phase, apoptosis, cell migration, and invasion were analyzed using transwell inserts, and EMT was quantified by the expression of mesenchymal and epithelial markers.
Results:
AMPK is activated in thyroid cancer cell lines, and AICAR treatment further increased AMPK phosphorylation. After 48 hours of AICAR treatment, the percentage of cells in the G2/M phase decreased, and a G0/G1-phase arrest was induced in both cell lines. AMPK activation effectively induced apoptosis in the BCPAP and TPC-1 cancer cell lines, while no apoptosis induction was observed in Nthy-ORI cells. AICAR also reduced the migration of Nthy-ORI and BCPAP cells by 30% and approximately 60% in TPC-1 cells. AICAR had no effect on cell invasion in Nthy-ORI and TPC-1 cells, but a significant reduction of cell invasion was observed in BCPAP cells. AICAR induced a significant reduction of N-cadherin and no changes in the expression of vimentin or TCF/Zeb1 protein in BCPAP cells. No differences in the expression of EMT markers were found in the AICAR-treated Nthy-ORI cells. A remarkable reduction of vimentin, TCF/Zeb1, and N-cadherin protein expression was detected in the TPC-1 cells.
Conclusions:
Increased activation of AMPK in PTC cell lines leads to a strong antitumor response, as measured by the inhibition of cell proliferation, cell migration, and induction of cell death. AMPK activation also reverses EMT in TPC-1 cells.
Introduction
D
In an effort to provide novel therapeutic approaches for patients with advanced DTC, two different tyrosine kinase inhibitors were approved for clinical use in advanced thyroid cancer patients in the last four years (8). Overall, the results of clinical studies have demonstrated a prolongation of the progression-free survival rate of the enrolled patients. However, the use of these drugs is associated with a relatively high rate of side effects, which reinforces the importance of identifying new molecular targets for thyroid cancer treatment.
It has recently been shown that the 5′-AMP-activated protein kinase (AMPK) pathway is overexpressed in PTC, the most common type of DTC, compared with non-tumor tissue from the same patient (9). It had previously been demonstrated that AMPK is expressed in normal rat thyroid glands and in the rat thyrocyte cell line PCCL3, regulating both glucose and iodine uptake (10 –12). These studies show that the pharmacological activation of AMPK by 5-aminoimidazole-4-carboxamide-ribonucleoside (AICAR) leads to increased glucose uptake, NIS protein degradation, and decreased iodide uptake by normal thyrocytes (10 –12).
Previous evidence suggests that the AMPK pathway is implicated in cellular and whole-body energy homeostasis, as well as in various aspects of tumor cell metabolism, proliferation, and metastasis (13,14). AMPK was initially described as a cellular energy sensor, monitoring changes in the intracellular AMP/ATP and ADP/ATP ratio in order to maintain energy homeostasis (15). It was also demonstrated that hypoxia, exercise, and nutrient deprivation activates the AMPK pathway in an attempt to stimulate energy-producing pathways and to inhibit energy-consuming pathways, with the goal of re-establishing intracellular ATP stores (13).
In addition to AMPK's role in the regulation of metabolism, the discovery that LKB1 is an upstream activator of AMPK raised the possibility that this enzyme could also be implicated in tumor biology (16 –18). In fact, a great body of evidence shows that multiple AMPK activator compounds such as metformin, phenformin, AICAR, and A769662 have tumor suppressor effects, inhibiting cell proliferation and inducing cell death in several types of tumors in vivo and in vitro (19 –27). However, under specific circumstances that seem to depend upon the developmental stage of the tumor, AMPK activation protects cancer cells from metabolic stress and improves tumor survival (28 –31). Thus, AMPK can exert a dual role in cancer, acting as either a tumor suppressor or a tumor promoter.
AMPK activation leads to reduced cell migration and invasion and reduced metastasis in experimental melanoma and breast cancer models (32 –34), and it has also been associated with the inhibition of epithelial-to-mesenchymal transition (EMT) (33,34).
In thyroid cancer cells, Choi et al. (22) demonstrated that AMPK activation by AICAR inhibits cell proliferation and stimulates apoptosis in papillary cancer cell lines harboring the BRAFV600E mutation, one of the most common mutations found in PTCs. The interplay between AMPK and mTOR in thyroid cancer is not well established, but previous studies have shown that cAMP inhibits the proliferation of TPC-1 cells through the downregulation of mTOR (35), a known target of AMPK. Epidemiological studies have reported that diabetic thyroid cancer patients treated with metformin, an oral anti-diabetic drug, show a reduced tumor size and higher remission rates (36). In addition, metformin has been shown to be anti-mitogenic and to induce cell death in PTC cells in vitro and in an in vivo xenograft model (37). Although several aspects of the mechanisms that regulate the effects of metformin are mediated by indirect AMPK activation, the pathophysiological role of AMPK in thyroid cancer remains unclear. Furthermore, the role of AMPK in regulating cell migration or invasion, as well as thyroid cancer progression, has never been investigated.
This study explored the effects AMPK activation induced by AICAR on different aspects of cancer cell behavior, including cell proliferation, apoptosis, cell migration, invasion, and EMT, in the human thyroid cancer cell line BCPAP that harbors the BRAFV600E mutation, as well as in TPC-1 cells, which harbor a RET/PTC translocation, thereby representing the most common genetic alterations found in PTC.
Materials and Methods
Reagents
AICAR was purchased from Toronto Research Chemicals, Inc. (Toronto, Canada). Polyvinylidene difluoride (PVDF) membranes and transwell inserts were purchased from Millipore (Billerica, MA). The ECL reagent and BCA protein assay kit were provided by Pierce (Rockford, IL). Specific antibodies against the total and phosphorylated forms of AMPK, vimentin, N-cadherin, and TCF/Zeb1 were purchased from Cell Signaling Technology, Inc. (Beverly, MA), and β-Actin antibody was purchased from Sigma-Aldrich (St. Louis, MO). The RPMI 1640 and Dulbecco's modified Eagle's medium (DMEM) were provided by HiMedia (Curitiba, Brazil), and fetal bovine serum (FBS) was from Life Technologies (Saint Aubin, France). The Matrigel matrix was purchased from BD Biosciences (Franklin Lakes, NJ).
Cell culture
The human thyroid epithelial non-tumor-derived cell line (Nthy-ORI) and the human thyroid papillary cancer cell lines (BCPAP and TPC-1) were generous gifts from Dr. Corinne Dupuy (Institut Gustave Roussy, Villejuif, France). The non-tumor-derived cell line Nthy-ORI was cultured in RPMI 1640 (HiMedia), and the cancer cell lines were grown in DMEM (HiMedia). Both media were supplemented with 10% FBS. Cells were maintained at 37°C in a 5% CO2–95% O2 mixture.
Cell viability
The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was used to test cell viability at different concentrations of AICAR treatment. Briefly, 1 × 104 cells were seeded in 96-well plates and cultured overnight. After that, cells were cultivated in the absence or presence of different concentrations of AICAR for 24 h, and the quantity of viable cells was measured by the absorbance at 450 nm using a VICTOR X4 Multilabel Plate Reader (Perkin Elmer, Waltham, MA).
ATP content
Cells were cultivated in 12-well plates. At 80% confluence, the cells were cultivated in the absence or presence of 1 mM of AICAR for 24 h. Next, the medium containing AICAR was removed, and the cells were washed twice and processed using a luciferin–luciferase bioluminescence assay kit, according to the instructions provided by the ADP/ATP ratio assay kit (MAK 135; Sigma-Aldrich). In the presence of luciferase, ATP immediately reacts with the Substrate D-luciferin to produce light. The light intensity is a direct measure of the intracellular ATP concentration.
Cell growth
Cells were seeded at a density of 103 cells per well in 96-well culture plates. After 24 h (time 0), the wells for each cell line were fixed with 100% methanol and incubated with the nuclear crystal violet dye. The dye was eluted in 100% methanol, and the absorbance was measured at 450 nm using a VICTOR X4 Multilabel Plate Reader (Perkin Elmer). Then, the Nthy-ORI, BCPAP, and TPC-1 cells were treated with 1 mM of AICAR for 24, 48, and 72 h. Untreated cells for each time point were used as the control. After the indicated periods of time, the quantity of viable cells was also determined by the crystal violet assay.
Western blotting
Cells were homogenized in lysis buffer containing 135 mM of NaCl, 1 mM of MgCl2, 2.7 mM of KCl, 20 mM of Tris, pH 8.0, 1% Triton, 10% glycerol, 0.5 mM of Na3VO4, 10 mM of NaF, and a protease and phosphatase inhibitor cocktail (Sigma-Aldrich). Subsequently, the lysates were centrifuged at 10,956 g and the supernatants were collected. An aliquot of the supernatant from each sample was used to determine the protein concentration using the BCA method (Thermo Fisher Scientific, Waltham, MA). Fifty micrograms of protein were then subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis, transferred to PVDF membranes (Millipore), and probed with specific antibodies against the total and phosphorylated forms of AMPK and acetyl-CoA carboxylase (ACC), vimentin, N-cadherin, or TCF/Zeb1 (Cell Signaling Technology, Inc.). β-Actin was used as loading control. Protein detection was performed using the enhanced chemiluminescence reagent obtained from Millipore and Pierce, and immuncomplexes were visualized using a C-DiGit® Blot Scanner (Li-Cor, Cambridge, United Kingdom).
Cell cycle assay
Cells were plated in 60 mm culture dishes (5 × 105 cells per dish) and treated with 1 mM of AICAR for 24, 48, or 72 h. The cell cycle phase was determined using the Muse Cell Cycle Kit (Millipore), following the manufacturer's instructions. Briefly, cells were trypsinized and centrifuged at 300 g for 5 min. The pellet was suspended in an ice-cold 70% ethanol solution and incubated at −20°C for 3 h. An aliquot was transferred to a new tube, centrifuged at 300 g for 5 min, and incubated with propidium iodide and RNAse A at room temperature for 30 min. The fluorescence intensity and cell cycle analysis were measured in a Muse Cell Analyzer (Millipore).
Apoptosis assay
Cells were plated in 60 mm culture dishes (5 × 105 cells per dish) and treated with 1 mM of AICAR for 24, 48, or 72 h. The cell apoptosis rate was measured using the Muse Annexin V & Dead Cell Kits (Millipore), according to the manufacturer's instructions. Briefly, cells were trypsinized and suspended in 1 mL of DMEM containing 10% FBS. A 200 μL aliquot was transferred to a new tube and mixed with 200 μL of the annexin V solution. After incubation for 20 min at room temperature, the fluorescence intensity was measured using a Muse Cell Analyzer (Millipore).
Transwell migration assay
Transwell inserts with 8 μm pore membrane filters were used for the migration assay, according to the manufacturer's protocol. Briefly, cells were cultivated in DMEM supplemented with 10% FBS in the presence or absence of 1 mM of AICAR for 16 h. Cells were then harvested by trypsinization and counted with Trypan Blue. A suspension of 5 × 104 cells in DMEM supplemented with 1% FBS was loaded into the upper chamber of each transwell in the presence or absence of 1 mM of AICAR. Next, 750 μL of the growth medium containing 5% FBS was loaded into the lower chamber. After an 8 h incubation in a humidified tissue culture incubator at 37°C and 5% CO2, the non-migrating cells were removed from the upper surface of the membrane by gently scrubbing with a cotton swab. The cells on the lower surface of the membrane were then fixed using 100% ethanol and stained with a 0.05% crystal violet solution followed by three washes with distilled water. The inserts were then allowed to air dry, and the membranes were photographed under an inverted microscope. The dye was then eluted with ethanol, and the absorbance was measured at 450 nm using a VICTOR X4 Multilabel Plate Reader. The data are expressed relative to the untreated control.
Cell invasion assay
The invasion assay was performed in 8 μm pore membrane transwell inserts coated with Matrigel (BD Biosciences, San Jose, CA). The Matrigel was diluted in a coating buffer (0.01 M of Tris, pH 8.0, 0.7% NaCl) to a final concentration of 250 μg/mL. Next, 100 μL of the Matrigel solution was added to the upper chamber of the transwell that was inserted into the wells of a 24-well plate and was incubated in a humidified tissue culture incubator at 37°C for 2 h. The cells were harvested by trypsinization, and a suspension of 5 × 104 cells in DMEM supplemented with 1% of FBS was loaded into the upper chamber of the transwell in the presence or absence of 1 mM of AICAR. The bottom chamber was loaded with 750 μL of DMEM containing 5% FBS. For each experimental group, the transwell inserts without the Matrigel coating were included as an internal control. After 24 h of incubation in a humidified tissue culture incubator at 37°C and 5% CO2, the non-invading cells were removed from the upper surface of the membrane by gently scrubbing with a cotton swab. Cells on the lower surface of the membrane were then fixed using ethanol and were stained with a 0.05% crystal violet solution followed by three washes with distilled water. The dye was then eluted with ethanol, and the absorbance was measured at 450 nm using a VICTOR X4 Multilabel Plate Reader.
For each experimental group, the ratio between the mean absorbance of the invading cells through the Matrigel-coated transwell membranes and the mean absorbance of the invading cells through the transwell membranes without the Matrigel coating was calculated. This ratio was multiplied by 100 in order to calculate the percentage of invasion, according to the manufacturer's protocol for the BD Matrigel matrix invasion chamber.
Statistical analysis
The data were analyzed using GraphPad Prism v5. The results are expressed as the mean ± standard error of the mean (SEM) of at least three independent experiments. For the cell growth curves, the cell cycle assay, and the apoptosis assays, the data were analyzed using a two-way analysis of variance, followed by a Bonferroni post hoc test. For the cell migration assay, the cell invasion assay, and the densitometry analysis of protein expression, the data were analyzed using a Student's t-test. Differences were considered to be statistically significant when p < 0.05.
Results
AMPK is constitutively activated in thyroid carcinoma cell lines
The cancer cell lines BCPAP and TPC-1 have a constitutively higher p-AMPK content than the non-tumor-derived Nthy-ORI thyroid cell line (Fig. 1). In order to validate these results, the expression of a well-known AMPK direct phosphorylation target, ACC, was analyzed. Higher basal levels of p-ACC in BCPAP and TPC1 cells compared to Nthy-ORI were also observed, which is an indicator of increased AMPK activation (Fig. 1).
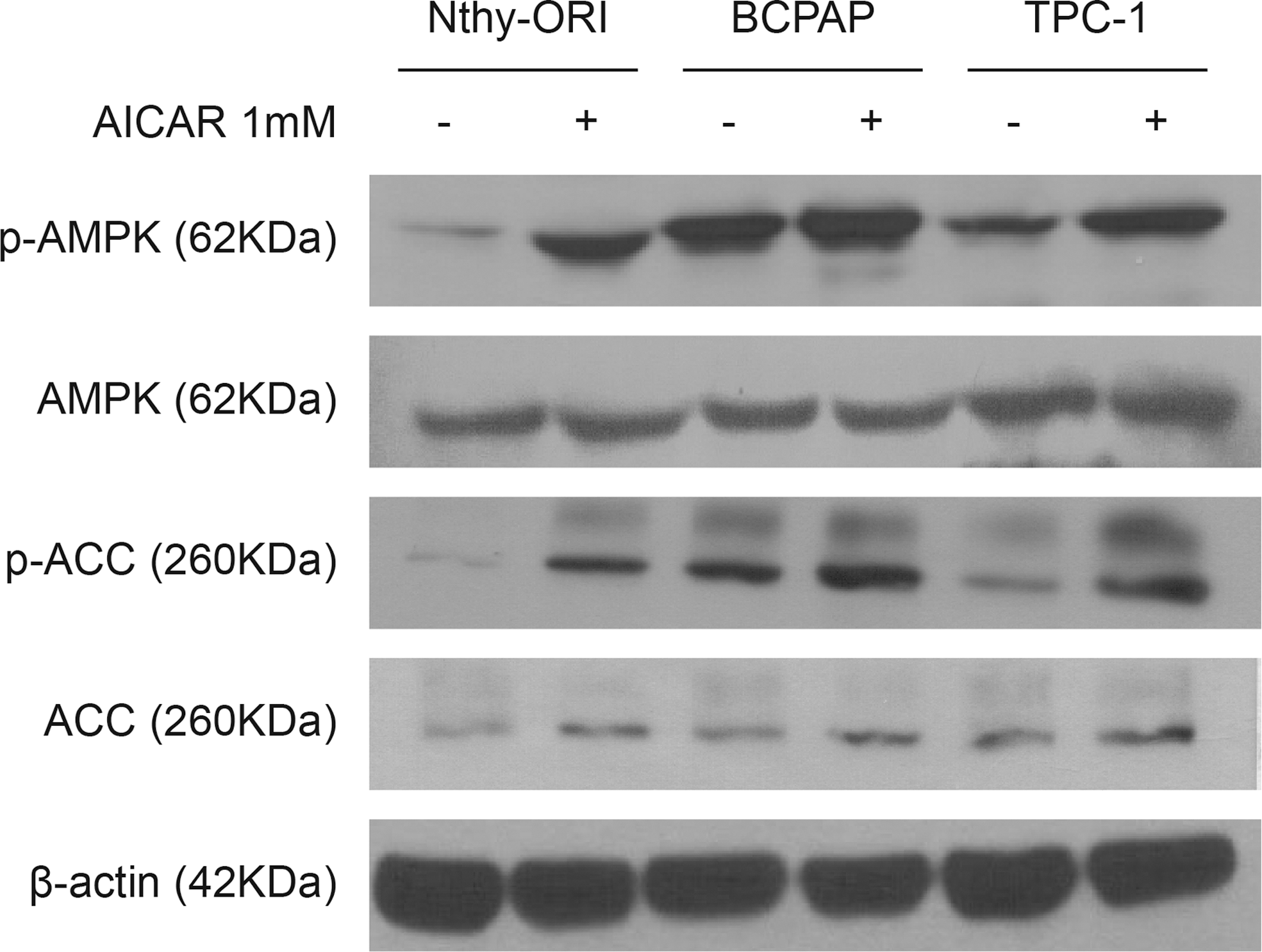
Effect of 5-aminoimidazole-4-carboxamide-ribonucleoside (AICAR) on 5′-AMP-activated protein kinase (AMPK) and acetyl-CoA carboxylase (ACC) protein expression. Total and phosphorylated AMPK and ACC protein expression were evaluated in Nthy-ORI, BCPAP, and TPC-1 cell lines in the presence (+) or absence (–) of 1 mM of AICAR for 24 h. β-Actin was used as loading control. The immunoblots shown are representative of three independent experiments.
Cells that were grown for 24 h in the presence of AICAR, the pharmacological activator of AMPK, showed a further increase of AMPK activation, as indicated by an increase of p-AMPK and p-ACC expression (Fig. 1).
AICAR treatment for 24 h significantly decreased the viability of the Nthy-ORI, BCPAP, and TPC-1 cells, beginning at doses of 0.5 mM in the TPC-1 cells, and 1.5 mM in the BCPAP and Nthy-ORI cells (Fig. 2A–C). Furthermore, treatment with 1 mM of AICAR led to increased intracellular ATP levels, and the majority of the cells were still viable at 24 h, confirming AMPK activation at this time point. Therefore, an intermediate dose of 1 mM of AICAR, which reduced cell viability, was chosen for further experiments (Fig. 2D–F).
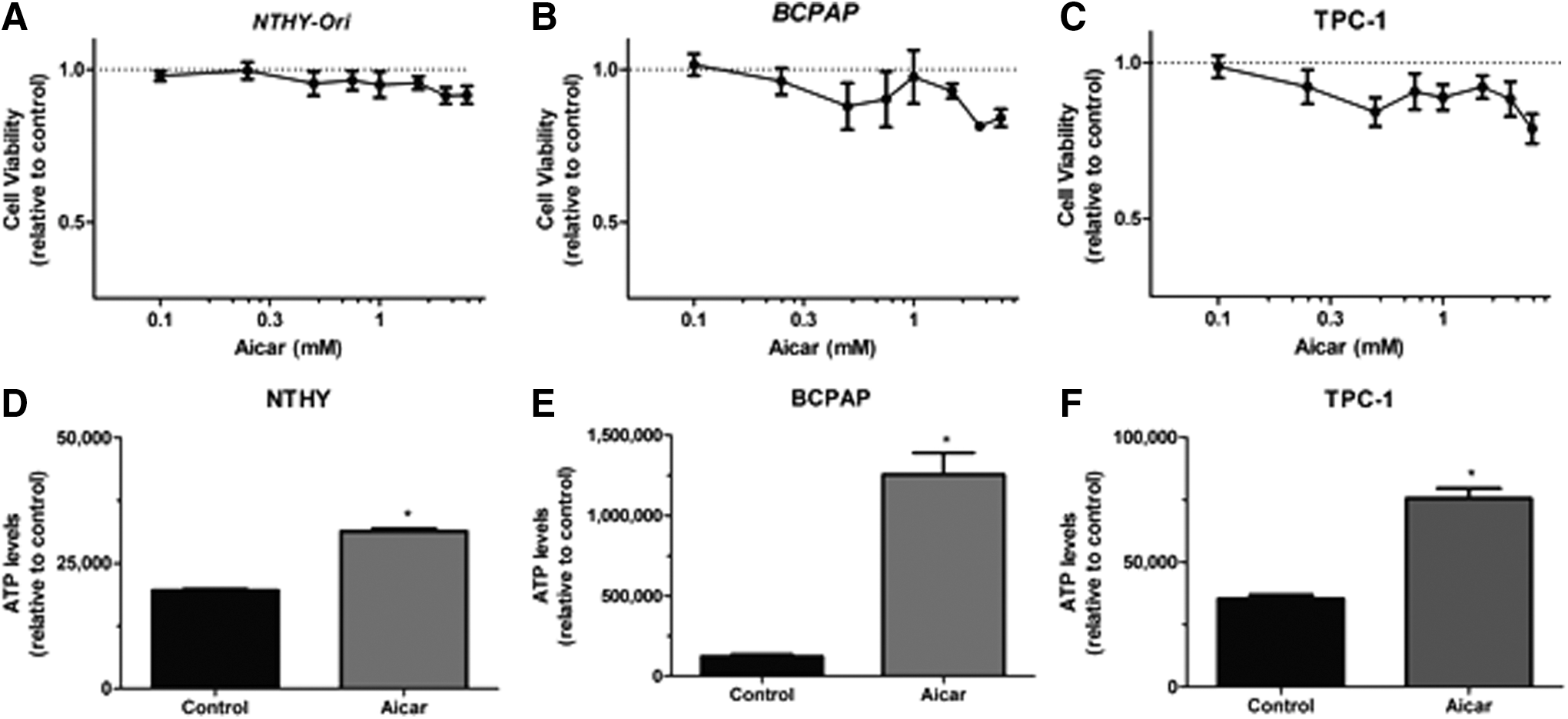
Effect of AICAR on cell viability and ATP content. Cells were treated in the absence or presence of different concentrations of AICAR for 24 h, and the number of viable (
AICAR inhibits cell proliferation of different thyroid cell lines
Because AMPK has been implicated in the regulation of cell proliferation in a wide variety of cancer cells, the effect of 1 mM of AICAR treatment was analyzed for up to 72 h on the cell proliferation of Nthy-ORI, BCPAP, and TPC-1 cells. The time-course analysis revealed that AICAR treatment for 48 h or 72 h inhibited cell growth in all of the cell lines (Fig. 3A–C). Then, the cell cycle progression was assessed, which revealed that AICAR treatment reduced the percentage of cells in the G2/M phase and induced a G0/G1-phase arrest in the Nthy-ORI (Fig. 4A) and the TPC-1 cells (Fig. 4C). In the TPC-1 cells, AICAR treatment for 24 h also increased the percentage of cells in the S phase compared to controls (Fig. 4C). In BCPAP cells, AICAR induced an increase in the S phase population at 48 h, with no further alterations in other cell cycle stages (Fig. 4B). At 72 h, an AICAR-induced reduction of the G2/M population and a G0–G1 phase arrest was observed (Fig. 4B).
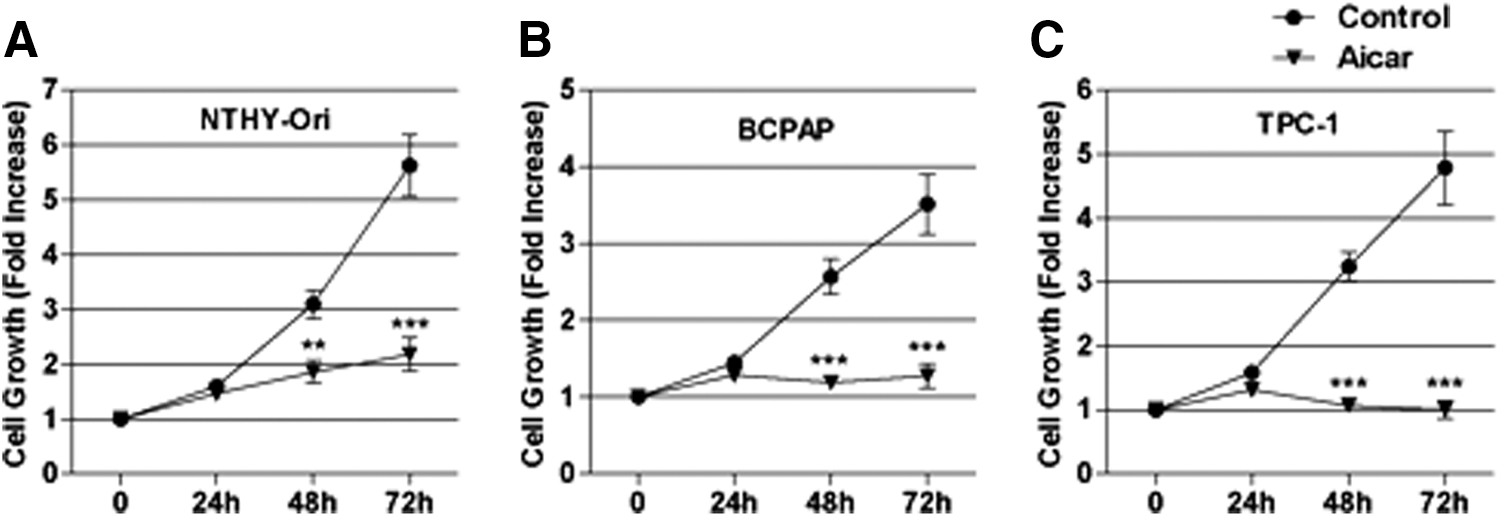
Effect of AICAR on cell growth. (
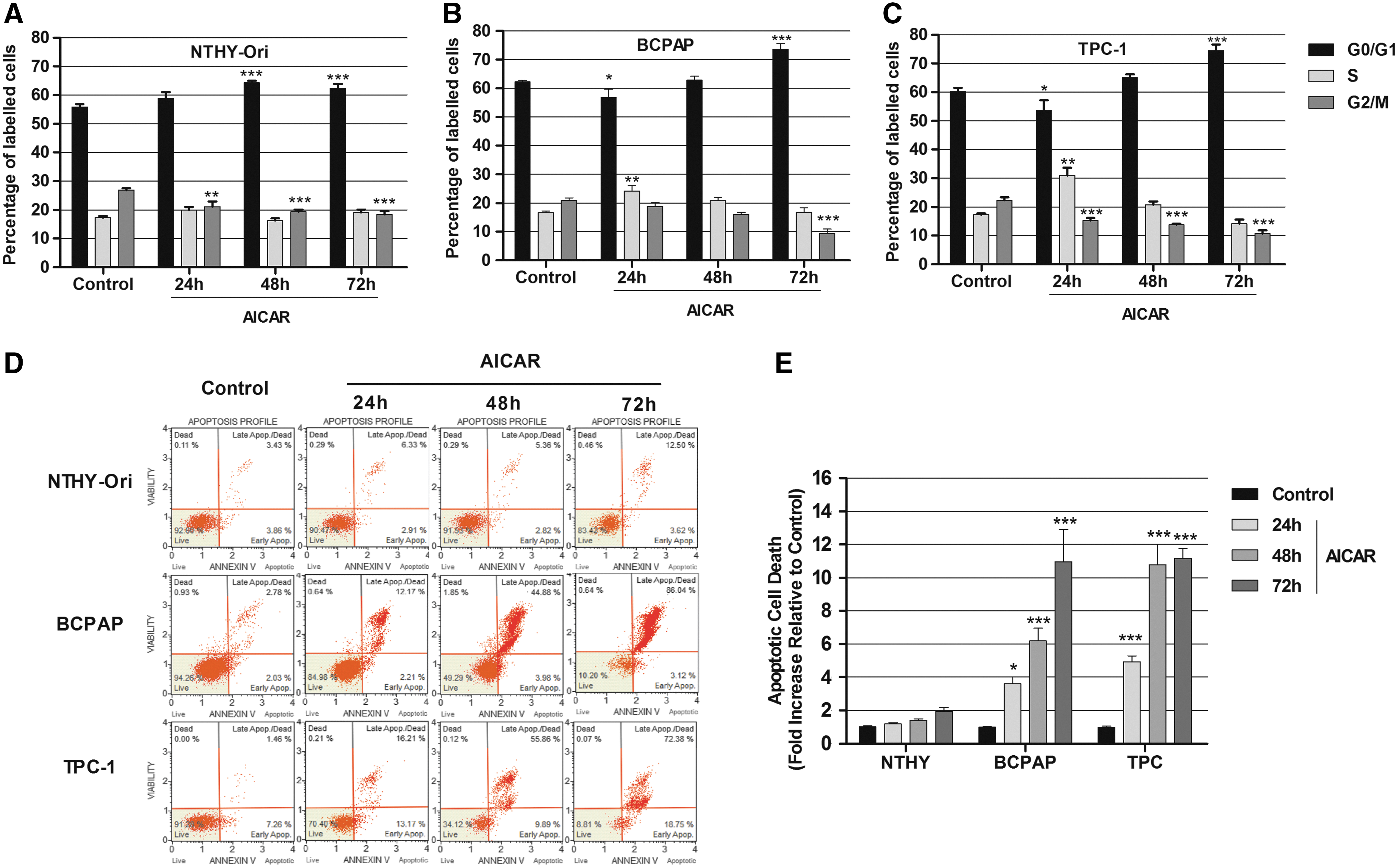
Effect of AICAR on the cell cycle and apoptotic cell death. Cells were treated in the absence or presence of 1 mM of AICAR for 24, 48, and 72 h. Cell cycle changes were measured by propidium iodide staining in (
PTC cells are susceptible to AICAR-induced apoptosis
This study further investigated whether the inhibition of cell growth could be attributed to the induction of apoptosis. The cells were treated with 1 mM of AICAR for 24, 48, and 72 h, and apoptotic cell death was assessed by annexin V staining (Fig. 4D and E). No differences were observed in the rates of apoptosis between controls and AICAR-treated Nthy-ORI cells. However, 24 h of AICAR treatment effectively induced apoptosis in the two cancer cell lines (Fig. 4D and E). The AICAR-induced pro-apoptotic response was further increased when the treatment was prolonged to 48 h or 72 h in the BCPAP and the TPC-1 cells (Fig. 4D and E). Interestingly, the TPC-1 cells seem to be more susceptible to AICAR-induced apoptosis than the BCPAP cells are because, after 48 h of AICAR treatment, a higher apoptotic rate was observed in the TPC-1 cells, while the quantity of apoptotic cells was similar in both of the tumor cell lines after 72 h.
AICAR inhibits Nthy-ORI and TPC-1 cell migration and reduces cell invasion of BCPAP cells
In an attempt to evaluate the effects of AICAR-induced AMPK activation on thyroid cell migration and invasion, the transwell migration/invasion assays were used. The cells were pretreated with AICAR for 16 h and plated onto the upper chamber of the transwell system in the presence or absence of AICAR for 8 h. Figure 5 shows representative micrographs of the cells migrating onto the lower surface of the transwell membrane stained with the crystal violet nuclear dye. It is notable that the density of purple staining is visually decreased in AICAR-treated groups compared with the respective untreated control in all of the cell lines tested, showing that the migratory ability of these cells is significantly reduced. AICAR caused a 30% reduction in the cell migration ability of the Nthy-ORI (Fig. 5A) and the BCPAP (Fig. 5B) cells. In the TPC-1 cells, there was a stronger effect of AICAR, reducing cell migration by approximately 60% compared with the respective control (Fig. 5C). In the invasion assay, however, no changes mediated by AICAR in the Nthy-ORI (Fig. 5D) and the TPC-1 cells were observed (Fig. 5F), but a significant reduction in cell invasion in the BCPAP cells was detected (Fig. 5E).
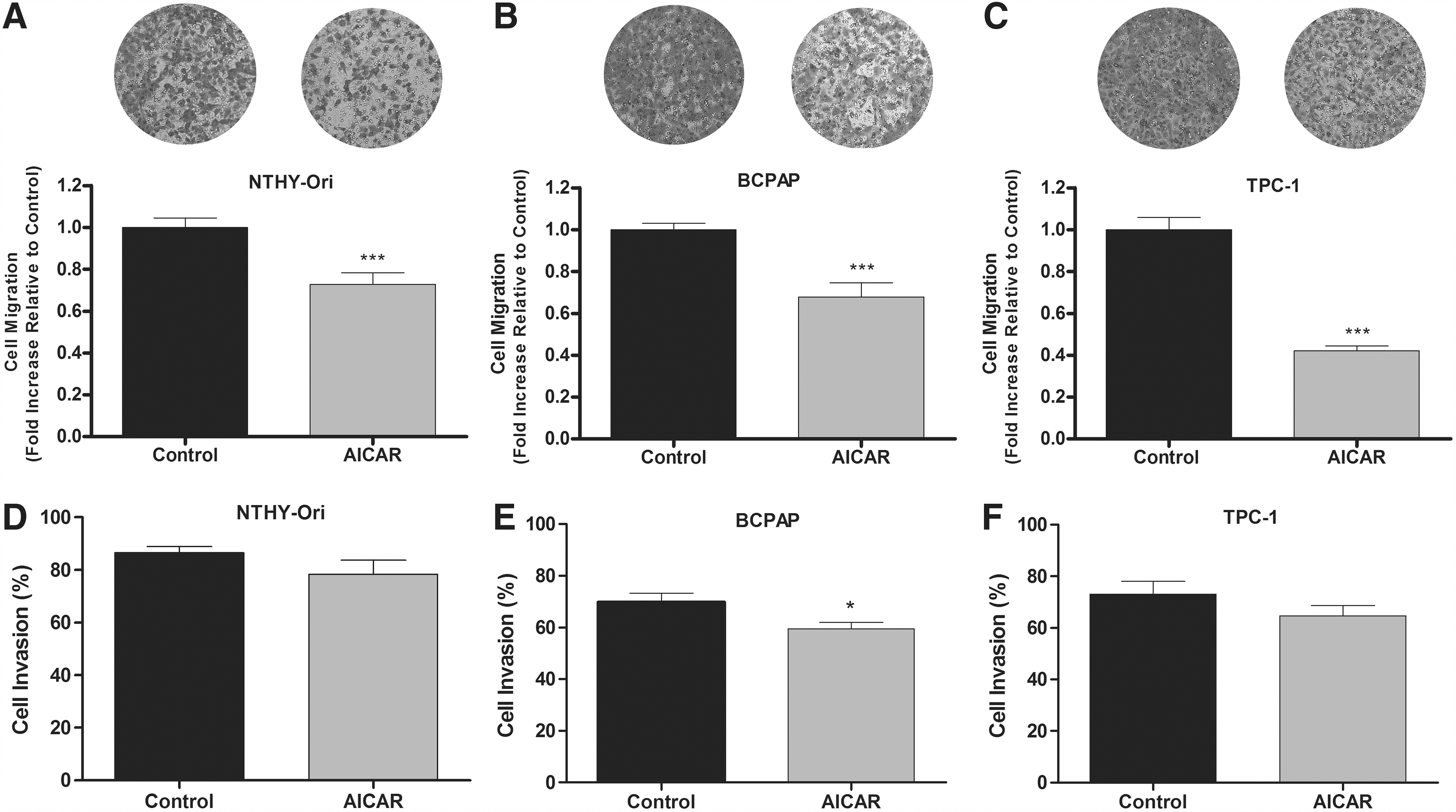
Effects of AICAR on cell migration and invasion in vitro. (
AICAR downregulates the expression of proteins involved in the EMT of PTC cell lines
Because the induction of EMT is associated with changes in cell survival, motility, and invasiveness, next this study evaluated whether AICAR is able to regulate EMT (Fig. 6). The cells were treated with AICAR for 24 h, and the expression of hallmark proteins associated with EMT was evaluated by Western blotting. There was no effect of AICAR treatment on the expression of EMT protein markers by the Nthy-ORI cells (Fig. 6A). In contrast, the AICAR-treated BCPAP cells had a significantly reduced N-cadherin protein expression (Fig. 6B), but no effect was observed in vimentin or TCF/Zeb1 expression (Fig. 6B). Interestingly, a remarkable effect of AICAR treatment on the TPC-1 cells was observed (Fig. 6C). Specifically, there was a significant reduction of vimentin, TCF/Zeb1, and N-cadherin expression (by approximately 60–70%), suggesting that AICAR inhibits EMT in this cell line (Fig. 6B).

The expression of epithelial-to-mesenchymal transition (EMT) markers. The total protein expression of the EMT markers TCF/Zeb1, N-cadherin, and vimentin is shown in (
Discussion
Over the last 10 years, AMPK has emerged as a potential target for cancer treatment, and several AMPK activator compounds have been shown to regulate cell survival and metastasis in a variety of cancer types. This study shows that AMPK activation by AICAR leads to an antitumor response, inhibiting cell proliferation, cell migration, and inducing cell death of the PTC cell lines harboring different driver mutations. It also demonstrates for the first time that AMPK activation suppresses the EMT in TPC-1 cells.
It has recently been shown that AMPK and its activated form p-AMPK are overexpressed in PTC compared with non-tumor tissue (9). Interestingly, in this in vitro study, it was observed that the BCPAP and the TPC-1 cancer cell lines show higher levels of p-AMPK expression than the non-tumor-derived Nthy-ORI cell line under basal conditions, as assessed by Western blotting (Fig. 1). This finding is in agreement with a previous report showing that AMPK is over-activated in the tumors of patients with PTC (9), as has been previously shown for the mTOR pathway (38).
Previous studies have shown that AMPK activation by metformin has anti-proliferative and pro-apoptotic effects in PTC and anaplastic cancer cells in vitro (37,39). Metformin also reduced tumor growth and increased the extension of necrotic areas in an ectopic tumor mouse model that was induced by the tumorigenic BHP10-3SC papillary carcinoma cell line (37). Furthermore, in diabetic patients with PTC, metformin treatment is associated with AMPK activation and reduced tumor size (35). It is notable that the mechanisms underlying the action of metformin have not been completely elucidated, but this drug indirectly activates AMPK in several cell types. Consistent with the involvement of AMPK in the effects of metformin, AICAR-induced AMPK activation has also been associated with anti-proliferative and pro-apoptotic responses in BRAFV600E -mutated thyroid cancer cell lines, a finding that was associated with the downregulation of the mTOR and MAPK pathways (22).
The present study confirms the data of Choi et al. (22), and demonstrates that AICAR inhibits cell growth and induces cell cycle arrest, not only in PTC cells harboring the BRAFV600E mutation, but also in the Nthy-ORI (a non-tumor-derived cell line) and the TPC-1 (harboring a RET/PTC translocation) cell lines (Figs. 3 and 4A–C). Furthermore, an AICAR-induced S phase cell cycle arrest was observed of BCPAP cells treated with AICAR. In addition, treatment with AICAR for 72 h led to a G0–G1 phase arrest and to the reduction of the cell population in G2/M. In the Nthy-ORI and TPC-1 cells, AICAR had an earlier effect, reducing the percentage of cells in the G2/M phase and inducing a G0/G1-phase arrest at 24 h.
This study also demonstrates that AICAR induces apoptosis in the BCPAP and TPC-1 cancer cell lines (Fig. 4D and E). After 48 h of AICAR treatment, the TPC-1 cells appear to be more sensitive to AICAR-induced apoptosis than the BCPAP cells. Interestingly, the apoptotic effect was not observed in the non-tumor-derived Nthy-ORI cell line.
Metastasis is responsible for 90% of the deaths in patients with solid tumors, and is a dynamic process that requires the invasion of the extracellular matrix, migration through the interstitial stroma, and penetration of blood or lymph vessels in order to reach a secondary site where the cells can settle and proliferate (40,41). In different cancer models, AMPK-activating drugs, such as AICAR, metformin, and OSU-53, were reported to inhibit cell migration and invasion in vitro and to reduce the formation of metastatic nodules in xenograft models (32,33,42). The present study investigated the role of AMPK activation on the migration and invasion of PTC cells in vitro. The results demonstrate that AICAR inhibits the cell migration of Nthy-ORI, BCPAP, and TPC-1 cells. AICAR also slightly reduced the invasiveness of the BCPAP cells, with no changes in this parameter observed in the other cell lines. Therefore, in addition to its role in cell proliferation and survival, AMPK also regulates cell migration in this model system.
EMT is a pathophysiological process that confers enhanced migratory and invasive properties to epithelial cells, which are central to metastasis. This process is characterized by the increased expression of mesenchymal markers (such as N-cadherin, vimentin, and Snail) and a loss of expression of epithelial markers (such as E-cadherin, claudin, and occludins) (43). In thyroid cancer, the loss of E-cadherin expression is a marker of the progression from DTC to undifferentiated carcinoma (44). In addition, Vasko et al. (45) showed that cells at the invasion front of PTC have alterations in their gene expression profiles that are consistent with EMT.
Recent evidence in the literature demonstrates that AMPK and its upstream kinase LKB1 play a key role in the suppression of EMT in a variety of cancer models, such as lung adenocarcinoma, melanoma, and aggressive breast and prostate cancer (32,43,44,46 –48). This study investigated for the first time whether AICAR-mediated AMPK activation is able to modulate EMT markers in thyroid cancer cells. The results do not show alterations in TCF/Zeb1, vimentin, and N-cadherin protein expression after 24 h of AICAR treatment in the Nthy-ORI cells, but in the BCPAP cells, AICAR induced a slight decrease in N-cadherin protein expression. In contrast, in the TPC-1 cells, a significant reduction in the expression of all of the studied EMT markers was observed, suggesting a more profound suppression of EMT in the TPC-1 cell line under these conditions.
It has previously been shown that thyrotropin (TSH) inhibits AMPK phosphorylation in normal rat thyroid cells (10). Because AMPK might act as an antitumor target, future studies should characterize the effects of TSH and AMPK in thyroid cancer cells, as well as the effect of TSH suppression, which is frequently required in the long-term therapy of patients with thyroid cancer.
In conclusion, this study shows that AMPK activation plays a major role in the induction of cytostatic and cytotoxic effects in two PTC cell lines. Furthermore, it shows that AMPK activation leads to an anti-migratory response and decreased expression of EMT markers in the TPC-1 cells. Taken together, these results suggest a potential antitumorigenic role of AMPK in PTC cell lines in vitro. More studies are necessary to address whether these effects are reproducible in animal models and humans, and in order to characterize the impact of other pathways influencing AMPK signaling, which may modulate the effects reported herein.
Footnotes
Acknowledgments
This work was supported by grants from the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Programa de Oncobiologia da UFRJ, Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ), and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES).
Author Disclosure Statement
The authors have nothing to declare.