
Abstract
Background:
Anaplastic thyroid carcinoma (ATC), the most aggressive type of thyroid cancer, has no effective therapy. Due to its dismal prognosis, it is vital to understand the genetic alterations of ATC and identify effective molecular targets. Targeted next-generation sequencing was performed to investigate the mutational profile of ATC using a massive parallel sequencing approach.
Methods:
DNA from formalin-fixed, paraffin-embedded archival samples of 11 ATCs and normal matched pairs were used. A total of 48 genetic alterations were identified by targeted exome sequencing. These alterations were validated by mass spectrometric genotyping and direct Sanger sequencing.
Results:
The most commonly mutated gene was BRAF, identified in 10 samples (91%), all showing the V600E point mutation. A KRAS point mutation was observed in the one sample (9%) without the BRAFV600E mutation. All 11 ATCs harbored BRAF or RAS mutations, reflecting the possibility that differentiated thyroid carcinomas progress to ATCs after the accumulation of mutations. A loss of function mutation of TP53 was observed in eight samples (73%), a PIK3CA mutation was observed in two samples (18%), and a frameshift mutation of PTEN was observed in one sample (9%). Twenty-eight novel mutated genes were found that had not previously been associated with ATC. Of these, loss of function mutations of NF2, KMT2D, and PKHD1 were repeatedly seen in three samples (27%), two samples (18%), and two samples (18%), respectively. Using direct Sanger sequencing, two samples (18%) were also found with a RASAL1 mutation. KMT2D and RASAL1 mutations were significantly associated with shorter ATC patient survival.
Conclusions:
This comprehensive analysis of ATCs using targeted massive parallel sequencing identified several novel mutations in ATCs, such as loss of function mutations of NF2 or KMT2D. Future studies are needed to confirm the role of these novel mutations as independent drivers of ATC development.
Introduction
A
Genetics studies of ATC have revealed frequent somatic mutations in BRAF and RAS, which are also commonly mutated in papillary thyroid cancer (PTC) and follicular thyroid cancer (FTC), as well as additional common PI3KCA, PTEN, TP53, CTNNB1, ALK, and TERT promoter mutations (2 –7). These findings suggest that ATC might develop from existing well-differentiated thyroid carcinoma with activation of both the mitogen activated protein kinase (MAPK) and phosphoinositide 3-kinase (PI3K)-AKT pathways by the accumulation of various genetic alterations. However, the precise molecular mechanisms involved in the dedifferentiation and progression of cancer are largely unknown. Recent advances in sequencing technologies might enable the comprehensive analysis of genetic alterations and identification of new targets.
In this study, targeted next-generation sequencing was performed to investigate the mutational profile of ATC using a massive parallel sequencing approach (8).
Materials and Methods
Patients and tissue samples
Eleven patients with ATC were enrolled who underwent thyroid surgery between 2009 and 2013 at the Asan Medical Center, Seoul, Korea. DNA from formalin-fixed, paraffin-embedded (FFPE) archival samples was used for targeted exome sequencing and validation. Only FFPE tissue samples from the initial thyroid surgery were used; no samples had been collected after radioactive iodine treatment. All pathological specimens were reviewed by an experienced pathologist (D.E.S.), and ATC diagnosis was confirmed according to the World Health Organization classification (9). The pathologist selected adequate tissue blocks for isolation of DNA from ATC tissues and matched normal tissues. The proportion of ATC cells in the FFPE tissues is indicated in Table 1. The protocol of this study was approved by the Institutional Review Board of the Asan Medical Center.
Age denotes age at diagnosis of ATC. N and M stages were classified by the 6th TNM staging system.
Proportion of ATC tumor in the FFPE tissue.
ATC, anaplastic thyroid carcinoma; FFPE, formalin-fixed, paraffin-embedded tissue; F, female; M, male; TT, total thyroidectomy; y, years; PTMC, papillary thyroid microcarcinoma.
DNA extraction methods
Matched hematoxylin and eosin–stained slides from each FFPE tissue were reviewed by a pathologist under the microscope. Genomic DNA was then extracted from between two and five 6-μm-thick sections per FFPE tissue; the number of sections used depended on the size and cellularity of the tumor. After de-paraffinization with xylene and ethanol, genomic DNA was isolated with a NEXprep FFPE Tissue Kit (#NexK-9000; Geneslabs, Seongnam, Korea) according to the manufacturer's recommendations. Briefly, tissue pellets were completely lysed by incubation with proteinase K in lysis buffer overnight at 56°C, followed by additional incubation for 3 min with magnetic beads and solution A at room temperature. After incubation for 5 min on a magnetic stand, the supernatant was removed and the beads were washed three times with ethanol. The beads were then dried for 5 min, DNA was eluted in 50 μL of DNase- and RNase-free water, and quantification was performed using a Quant-iT™ PicoGreen dsDNA Assay kit (Invitrogen, Carlsbad, CA).
Targeted next-generation sequencing
For evaluation of the somatic mutations in the 11 ATC tissues, targeted next-generation sequencing was performed using the MiSeq platform (Illumina, Inc., San Diego, CA) with OncoPanel version 2 (OP_v2) to capture the exons of 505 cancer-related genes plus partial introns from 15 genes often rearranged in cancer (Supplementary Table S1; Supplementary Data are available online at
Sequenced reads were aligned to the human reference genome (NCBI build 37) with Burrows-Wheeler Aligner (0.5.9) (13) with default options, and de-multiplexing was performed with MarkDuplicates of the Picard package to remove PCR duplicates. De-duplicated reads were realigned at known indel positions with GATK IndelRealigner (14), then base quality was recalibrated using GATK TableRecalibration. Somatic variant calling for single nucleotide variants and short indels was performed with matched normal tissue using MuTect (1.1.7) (15) and SomaticIndelocator in GATK, respectively. Germline variants from candidates of somatic variants were filtered out with common dbsnp (build 141; found in ≥1% of samples) and a panel of normal samples. Final somatic variants were annotated using Variant Effect Predictor (v79) and were then converted to maf file format using vcf2maf. Structural variations were identified using BreaKmer and then were filtered out with a panel of normal samples (16).
Validation of mutations using MassARRAY
To validate genetic alterations detected by targeted exome sequencing, mass spectrometric genotyping was performed using the Sequenom MassARRAY technology platform (Sequenom, San Diego, CA). Four different multiplex amplification pools for 45 mutation candidates were designed by the Assay Designer unit of the MassARRAY TYPER software (Supplementary Table S3). While the detection limit of mutant alleles varies between assays and samples, it is usually considered to be 5–10% (17). After multiplex PCR, treatment with shrimp alkaline phosphatase from the iPLEX-Pro kit (10142-2; Sequenom) was done to inactivate the residual deoxynucleotides, which were then subjected to single-base extension. After spotting of the desalted product onto a 384-format SpectroCHIP II, the spectrum profiles generated by matrix-assisted laser desorption/ionization time of flight mass spectrometry were acquired and interpreted using the TYPER 4.0 software (Sequenom), and then further reviewed manually by two independent researchers to undo any uncertain calls resulting from clustering artifacts.
Sanger sequencing validation
Seven somatic mutation candidates for the KMT2D, NF2, and PKHD1 genes were additionally validated by direct Sanger sequencing. Mutations in RASAL1, which was recently defined as a candidate driver gene of ATC, were also additionally evaluated by direct Sanger sequencing using the 11 ATC tissues (12). DNA was PCR amplified using a 2720 Thermal Cycler (Applied Biosystems, Foster City, CA). The 20 μL reaction volume contained a 250 μM mixture of dATP, dGTP, dTTP, and dCTP, 0.25 IU of AmpliTaq-Gold DNA polymerase, and 0.5 μM of each primer pair shown in Supplementary Table S4. The amplification protocol consisted of initial denaturation at 95°C for 15 min, followed by 45 cycles of denaturation at 95°C for 30 sec, annealing at 60°C for 30 sec, extension at 72°C for 30 sec, and a final extension at 72°C for 5 min. Amplified PCR products were sequenced using the forward or reverse PCR primers and Big Dye Terminator v3.0 cycle sequencing reagents (Applied Biosystems). PCR amplification consisted of 25 cycles of denaturation at 96°C for 10 sec, annealing at 50°C for 5 sec, and extension at 60°C for 4 min. Each DNA sequence was read on an ABIPRISM 3100 automated sequencer (Applied Biosystems).
Statistical analysis
Survival curves were constructed using the Kaplan–Meier method, and the log-rank test was used to evaluate differences in survival according to genetic alterations. All p-values were two sided, with p < 0.05 considered statistically significant.
Results
Clinical and pathological characteristics of the ATC patients
The clinical and pathological characteristics of the ATC patients in this study are presented in Table 1. The mean age of the 11 patients at thyroid surgery was 74.4 years (median 75 years), and eight patients (73%) were women. Nine patients underwent total thyroidectomy, and two patients underwent debulking surgery. The mean maximal diameter of the ATC was 4.5 cm (median 4.5 cm), and distant metastasis of the ATC was confirmed in six patients (55%) at initial diagnosis. Papillary microcarcinomas in the contralateral thyroid lobe were incidentally found in two patients. All patients died during follow-up, and the median survival time was 2.8 months.
Targeted next-generation sequencing
A total of 461 somatic variants were identified, and 76 of them were non-synonymous mutations: 43 coding missense mutations, 18 frameshift mutations, 10 non-sense mutations, 4 mutations at splicing sites, and 1 deletion. The overall mean target coverage, on target rate, and percentage of bases covered >20 times for the 11 ATC tumors were 147 reads, 36%, and 96%, respectively.
Among the 76 non-synonymous mutations, 28 were manually filtered out because of the low depth of each variant allele (the thresholds for MuTect and Somatic Indelocator were 5 and 10 counts, respectively), and the remaining 48 variations were considered true positive. These 48 variations were validated using the Sequenom MassARRAY system, and all mutations except one were confirmed. The mutant allele frequency of the unconfirmed genetic variation in targeted exome sequencing was 8%, close to the borderline of the detection limit of the MassARRAY system. The results of the targeted exome sequencing validated by the MassARRAY system are detailed in Figure 1 and Table 2.
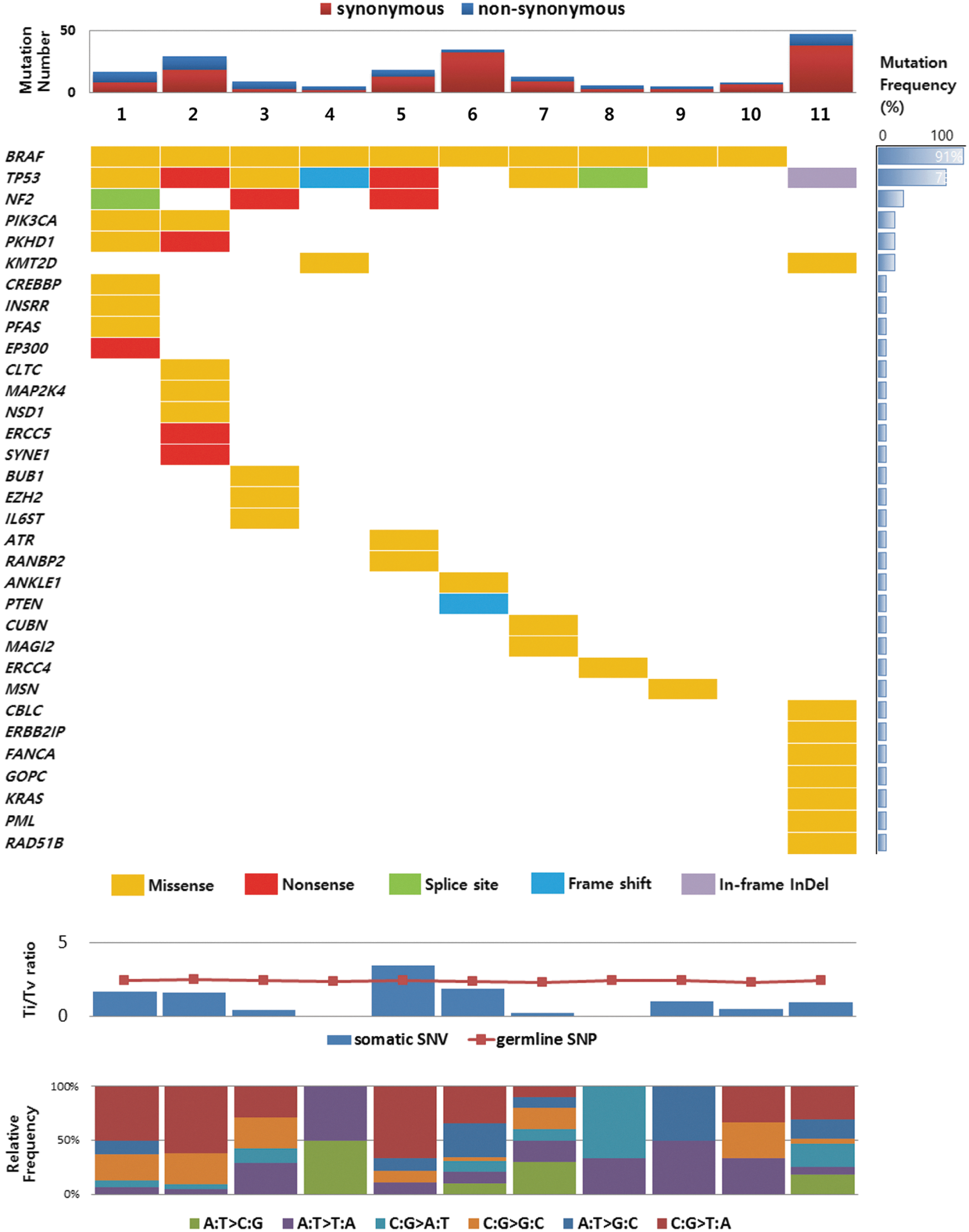
Somatic mutations in anaplastic thyroid carcinomas (ATCs) detected by targeted massive parallel sequencing. Tumor samples are arranged from left to right. Somatic gene mutations found in 11 ATCs are annotated for each sample according to the color panel below the image. The somatic mutation frequencies for each mutated gene are plotted in the right panel. The mutation number, transition/transversion ratio, and type of base-pair substitution are displayed in the top, middle, and bottom panels, respectively.
These three different mutations in PIK3CA were detected in one sample.
ATC-related gene mutations
The top 20 recurrently mutated genes were defined according to the COSMIC database of ATC as the ATC-related genes (18,19). All 11 ATCs had ATC-related gene mutations, such as BRAF, KRAS, TP53, PIK3CA, and PTEN mutations (Fig. 1 and Table 2). The most commonly mutated gene was BRAF, seen in 10 samples (91%), all showing the V600E point mutation. The KRAS point mutation was observed in the one sample (9%) without the BRAFV600E mutation. A loss of function mutation of TP53 was observed in eight samples (73%): three missense mutations, two non-sense mutations, one frameshift mutation, one deletion, and one mutation at a splicing site. PIK3CA missense mutations were observed in two samples (18%). A frame shift deletion mutation of PTEN was observed in the one sample (9%) Two samples did not harbor additional ATC-related gene mutations other than the BRAFV600E mutation.
Novel mutated genes in ATC
There were 32 somatic mutations in 28 novel genes in the 11 ATCs (Fig. 1 and Table 2). Of the 28 novel
All novel somatic mutations in the NF2, KMT2D, and PKHD1 genes were confirmed by direct Sanger sequencing (Supplementary Fig. S1). Additionally, RASAL1 mutations were evaluated by direct Sanger sequencing, and two missense mutations were detected among the 11 ATCs (18%; patients 1 and 11). These alterations were all located in the RAS GTPase-activating domain, as previously reported (12) (Supplementary Fig. S1).
Survival differences according to genetic alterations
Survival curves were constructed, and the differences were compared according to the mutations in the BRAF, TP53, NF2, KMT2D, PKHD1, and RASAL1 genes. The presence of a KMT2D or RASAL1 mutation was significantly associated with shorter ATC patient survival (p = 0.004 and p = 0.031, respectively). The presence of a TP53 or NF2 mutation also tended to affect the survival curves, but the results were not statistically significant (Fig. 2).
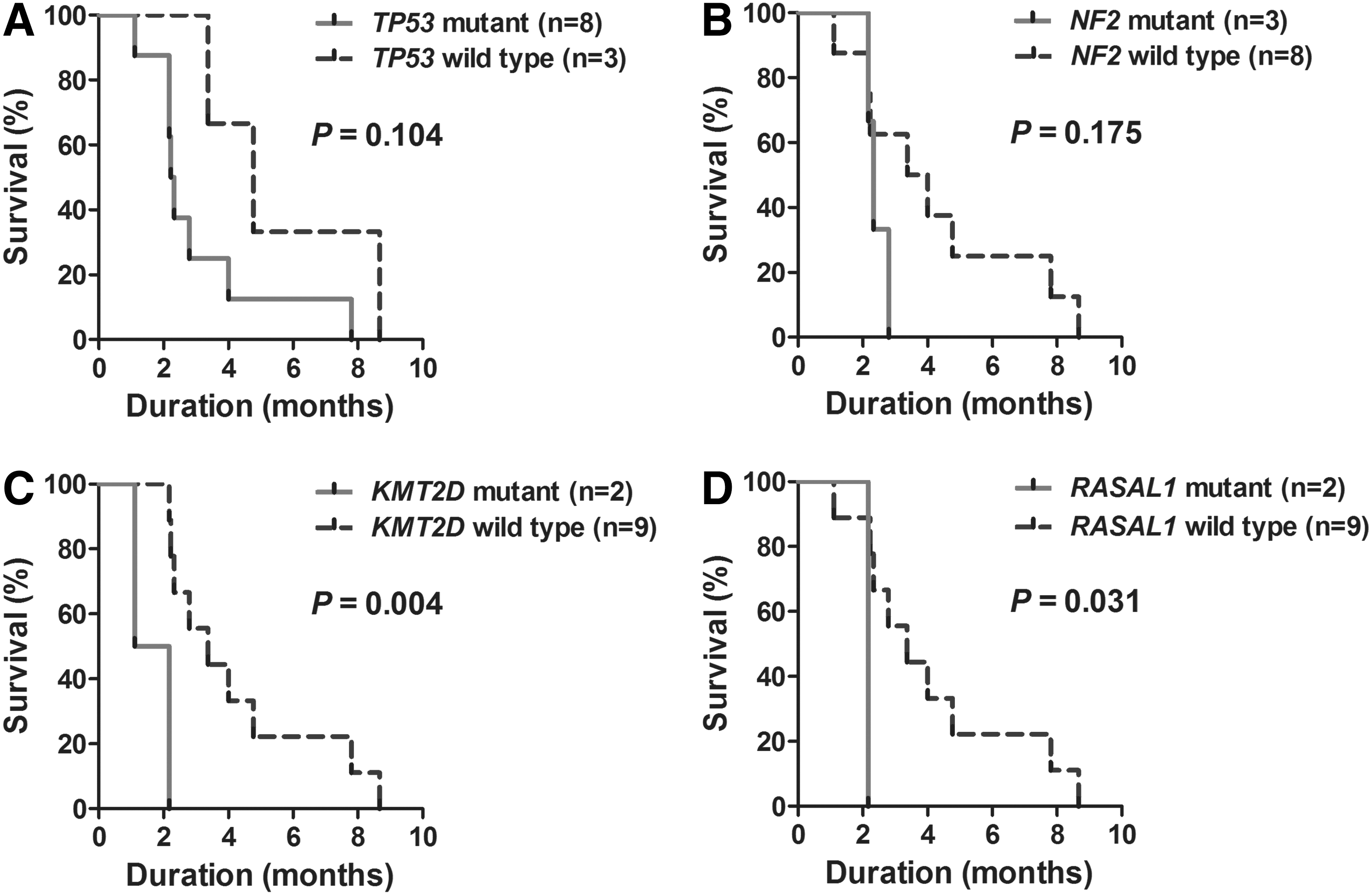
Survival curves of patients with anaplastic thyroid carcinomas according to genetic alterations. Presence of KMT2D and RASAL1 mutations was significantly associated with shorter survival of anaplastic thyroid carcinoma patients. (
Gene rearrangements in ATC
The presence of rearrangements were tested in 15 genes, including RET, ALK, BRAF, and AKT. However, there were no gene rearrangements in the 11 ATCs.
Comparison of the types of nucleotide substitutions between somatic and germline mutants
In the current study of 11 ATC cases, the transition (Ti)/transversion (Tv) ratio of germline single nucleotide polymorphisms (SNPs) was 2.41 (19,923/8264). However, in the case of somatic single nucleotide variants, the Ti/Tv ratio was dramatically decreased to 1.10 (76/69; Fig. 1).
Discussion
Targeted next-generation sequencing was performed using a massive parallel screening method. With this up-to-date technique, five ATC-related gene mutations and 28 novel gene mutations were detected in 11 ATCs. These data confirm the heterogeneous genetic background of ATCs.
The most frequently observed mutation in the ATCs was the BRAFV600E mutation, with a prevalence of 91%. This prevalence is markedly higher than that of previous reports, which indicated that BRAF mutations occur in about 10–38% of ATCs (Table 3) (22 –24). The reason for this difference is unknown. However, the prevalence of the BRAFV600E mutation of PTC in Korea has been reported to be higher than in other geographic regions, possibly due to differences in genetic backgrounds or iodine intake (25,26). All BRAF mutations were mutually exclusive with the RAS mutation, and only one KRAS codon 12 mutation was observed in this study. All 11 ATCs harbored a BRAF or RAS mutation, suggesting that PTC or FTC might progress to ATC with the accumulation of mutations. The additional loss of function mutation of TP53 is one of the mechanisms promoting the dedifferentiation of well-differentiated thyroid cancer (5,27), and it was found in 73% of the current ATC cases. Additional PI3KCA and PTEN mutations were observed in 18% and 9% of the ATCs, respectively. However, there were no additional mutations of known ATC-related genes other than BRAF or RAS in two ATCs, and the dedifferentiation mechanisms in these cases are uncertain.
A loss of function mutation of the NF2 gene was the most frequent novel mutation in the 11 ATC patients (3/11; 27%). The NF2 mutation is associated with neurofibromatosis, acoustic neuroma, and meningioma, as well as breast, colorectal, hepatic, and prostate cancer (28). NF2 is a tumor suppressor gene that encodes the protein merlin, which acts as a scaffold protein that modulates receptor-mediated signaling pathways such as receptor tyrosine kinase, small GTPase, PI3K/AKT/mTOR (mammalian target of rapamycin), and hippo pathways (28). One previous study reported a frequent allelic loss of NF2 in aggressive medullary thyroid carcinomas (29), and another recent study that performed whole exome sequencing of ATCs also reported that mutations in NF2 genes were novel driver mutations in ATCs (18). Furthermore, one recent study reported that NF2 inactivation increased MAPK output by promoting RAS transcription (30).
Mutations in KMT2D were also repeatedly observed in the ATCs in the present study. KMT2D, formerly named MLL2, is a histone methyltransferase that regulates gene transcription by modulating chromatin accessibility. KMT2D is frequently mutated in medulloblastoma, renal, bladder, gastric, hepatic, colorectal, and lung cancer, and previous studies have indicated that KMT2D is a tumor suppressor (31 –34). To the authors' knowledge, the current study is the first to report KMT2D mutations in thyroid cancer. Furthermore, patients with ATCs harboring KMT2D mutations had significantly shorter survival than those with ATCs without KMT2D mutations, suggesting a possible prognostic role of KMT2D mutations in ATC patients. Further studies are needed to uncover the detailed molecular mechanisms how loss of function mutations in NF2 or KMT2D affect the pathogenesis of ATC.
Inactivation of RASAL1, which encodes a RAS GTPase-activating protein, may promote ATC development by upregulating the PI3K pathway (12). The present study also found two samples with a RASAL1 mutation among the 11 ATCs by direct Sanger sequencing. These findings confirm the importance of the RASAL1 mutation in the pathogenesis of ATC. RASAL1 mutations were also associated with shorter patient survival in the present analysis. Further studies are needed to determine the prognostic role of RASAL1 mutations in ATC patients.
One limitation of the current study is that it involved a small series of 11 ATCs. Furthermore, not all mutated genes could be detected because this panel included just 505 known cancer-related genes. However, the number of subjects was comparable to previous studies of ATC because of its extremely low prevalence (6,35), and the present study is noteworthy because comprehensive analysis of ATCs was performed using targeted next-generation sequencing analysis with the MassARRAY process validation. Genetic changes detected as germline mutations were also excluded by comparing the results of the mutational profile in the ATCs with those of matched normal tissues. Significant differences in the Ti/Tv ratio between germline mutations (2.41) and somatic mutations (1.10) in the current study series confirmed the adequacy of the sequencing analysis. With regard to single nucleotide substitutions, all substitutions can be classified by six patterns of base substitutions: two types of Ti (C:G>T:A, A:T>G:C) and four types of Tv (C:G>A:T, C:G>G:C, A:T>C:G, A:T>T:A). During evolution, Ti are much more frequent than Tv, and the number of Ti over the number of Tv, referred to as the Ti/Tv ratio, in known SNPs is expected to be between two and four (36,37).
In conclusion, this comprehensive analysis of ATC using targeted massive parallel sequencing identified several novel mutations in ATCs, such as loss of function mutations in NF2 and KMT2D. Future studies are needed to confirm the role of these mutation
Footnotes
Acknowledgments
This study was supported by the National Research Foundation (NRF) of Korea Research Grant (NRF-2015R1C1A1A02036597).
Author Disclosure Statement
The authors have no competing interests to disclose.