
Abstract
Background:
Familial non-medullary thyroid cancer (NMTC) occurs either as part of known hereditary syndromes or as a non-syndromic isolated hereditary tumor. Although the genes underlying the syndromic type of NMTC have been identified in most syndromes, no clear underlying gene has been identified in the non-syndromic NMTC. Recently, a c.1601G>A, p.G534E mutation in the HABP2 gene was reported to be the underlying genetic defect in a family with seven members affected by NMTC. The G534E variant has also been reported to occur in about 4.7% of cases of the Thyroid Cancer Genome Atlas (TCGA) database.
Objectives:
The aim of this study was to explore whether the recent finding of G534E genetic variant can be replicated in a large sample of NMTC, including 11 members of four unrelated families with familial NMTC and 509 cases of sporadic pediatric (63 cases) and adult NMTC (446 cases).
Methods:
All exons and exon-intron boundaries of HABP2 were screened in 11 members of four families with familial non-syndromic NMTC using DNA isolated from peripheral leucocytes, polymerase chain reaction, and direct sequencing. The G534E variant was also screened for specifically in 229 cases of sporadic NMTC using DNA isolated from peripheral leucocytes and an additional 217 cases of NMTC using DNA isolated from formalin-fixed paraffin-embedded tumor tissues. As a control cohort, 190 healthy individuals without known thyroid disease were also studied for the presence of the G534E variant using DNA isolated from peripheral leucocytes.
Results:
None of the familial NMTC carried HABP2 mutations. Of 509 sporadic NMTC, only one case (0.2%) harbored the G534E variant. Similarly, only one case (0.5%) of the control group harbored the G534E variant.
Conclusion:
In this study, HABP2 mutations were not found in familial NMTC, and the G534E variant is not the underlying genetic defect in a large sample of sporadic NMTC from the Middle East.
Introduction
T
The population in the present study is highly inbred with a high rate of consanguinity, and TC is very common (12 –14). TC is the second most common cancer in females and the sixth most common cancer in Saudi Arabia (15). Therefore, it became intriguing to ask the question whether this recently described mutation in HABP2 gene is the underlying genetic defect in familial cases of TC and in some cases of the apparently sporadic NMTC in this population, especially in pediatric TC cases, since they are more likely to have a genetic predisposition for early cancer development. Therefore, a large number of patients with follicular cell-derived TC including familial cases and sporadic pediatric and adult cases were studied for mutations in HABP2. The results strongly suggest that the HABP2 G534E mutation is not a common underlying genetic defect and HABP2 does not seem to be an important underlying gene in NMTC, at least in this population.
Patients and Methods
Patients
The study was undertaken after obtaining Institutional Review Board approval and an informed consent when appropriate. The approach was as follows: we screened all exons and exon-intron boundaries of HABP2 gene for mutations in 11 members from 4 unrelated families. Each family had two or more members diagnosed with NMTC (Table 1). Only one patient was male. All patients were young (<45 years of age). None of the families had a history of another cancer or features suggestive of a specific syndrome (e.g., Cowden syndrome) that is known to be associated with TC. Genomic DNA isolated from peripheral leucocytes was used in these families. All 13 exons and the exon–intron boundaries of the HABP2 gene were screened using primers and polymerase chain reaction (PCR) conditions, as described before (6).
CPTC, conventional papillary thyroid cancer; LN, lymph node; NMTC, non-medullary thyroid cancer; TC-PTC, tall-cell variant papillary thyroid cancer.
Next, the focus was on the G534E mutation in the HABP2 gene located in exon 13. The study began with 63 pediatric patients (median age = 16 years, range 9–18 years), and the search was extended to another 229 adult cases of NMTC (median age = 40 years, range 21–75 years). In the pediatric and adult cases of NMTC, DNA was isolated from formalin-fixed paraffin-embedded tumor tissue and exon 13 was amplified by PCR using primers and PCR conditions previously described (6). To increase the cohort, another random sample of 217 sporadic NMTC cases were then added using peripheral leucocytes for DNA isolation and the same PCR and sequencing conditions. Finally, as a control, 190 healthy subjects with no known thyroid disease were screened for the same mutation using DNA isolated from peripheral leucocytes and PCR and sequencing conditions as above. Thus, in total, 710 cases were screened: four families (11 members) with mutational analysis of all exons of the HABP2 gene, and analysis for the presence of the G534E variant in exon 13 in a total of 63 pediatric and 229 adult cases of NMTC using tumor DNA, in 217 cases of NMTC using genomic DNA from peripheral leucocytes, and in 190 normal control subjects using DNA from peripheral leucocytes. Thus, the total number of sporadic NMTC consisted of 509 subjects, the number of familial cases was 11, and 190 were normal control subjects.
The clinical and pathological features of the familial cases are summarized in Table 1. The sporadic TC patients comprised 152 males (29.9%) and 357 females (70.1%). Their median age at diagnosis was 34 years (range 9–75 years). The tumor types were: 321 (63%) PTC, 96 (18.9%) follicular variant PTC, 32 (6.3%) tall-cell variant, 24 (4.7%) follicular thyroid cancer, 11 (2.2%) Hürthle cell cancer, 19 (3.7%) poorly differentiated thyroid cancer, and 6 (1.2%) anaplastic thyroid cancer. The median tumor size was 3.2 cm (range 0.7–13 cm). Tumor multifocality was present in 178 (35%) of cases, extrathyroidal extension in 213 (41.8%), lymphovascualr invasion in 92 (18%), lymph node metastasis in 222 (43.6%), and distant metastasis in 42 (8.3%) cases. The control sample comprised 190 cases (57 males), with a median age of 38 years (range 17–67 years).
DNA isolation, PCR, and direct sequencing
Genomic DNA from peripheral blood leucocytes was isolated using the Gentra Blood Kit (Qiagen Corp, Valencia, CA) according to the manufacturer's instructions.
For tumor tissue, the pathological specimen from each patient was examined by an experienced endocrine pathologist (H.A.) and tumor tissue was carefully dissected from formalin-fixed paraffin-embedded tissue blocks. DNA was extracted using a commercial DNA extraction kit (QIAamp DNA FFPE Tissue Kit; QIAGEN, Hilden, Germany; catalog no. 56404) according to the manufacturer's instructions. PCR and direct sequencing using Big Dye terminator v3.1 cycle sequencing reaction kit and an ABI PRISM 3730X genetic analyzer (Applied Biosystems, Foster City, CA) were used to detect mutations in HABP2 using the same primers and PCR conditions as previously described (6).
Results
HABP2 in familial NMTC
In the four families with NMTC, none of the 11 members carried the HABP2 G534E mutation. In these families, DNA isolated from blood samples was used. In one of these patients (family #2, daughter), the tumor DNA was also screened for the HABP2 G534E mutation, and it too was negative. Furthermore, mutational analysis of all 13 exons and exon–intron boundaries of the HABP2 gene in the index cases of these families did not reveal any mutation.
HABP2 G534E mutation in pediatric and adult TC using tumor DNA
None of the 63 pediatric TC cases harbored the G534E mutation. Only one patient among the 229 adult NMTC cases carried a monoallelic G534E mutation (Fig. 1). The patient was a 33-year-old female with a 4 cm conventional multifocal PTC with extrathyroidal invasion and lymph node metastasis. In addition to PTC, there was evidence of multinodular goiter in the rest of the thyroid gland. She had no family history of thyroid cancer or other thyroid diseases.
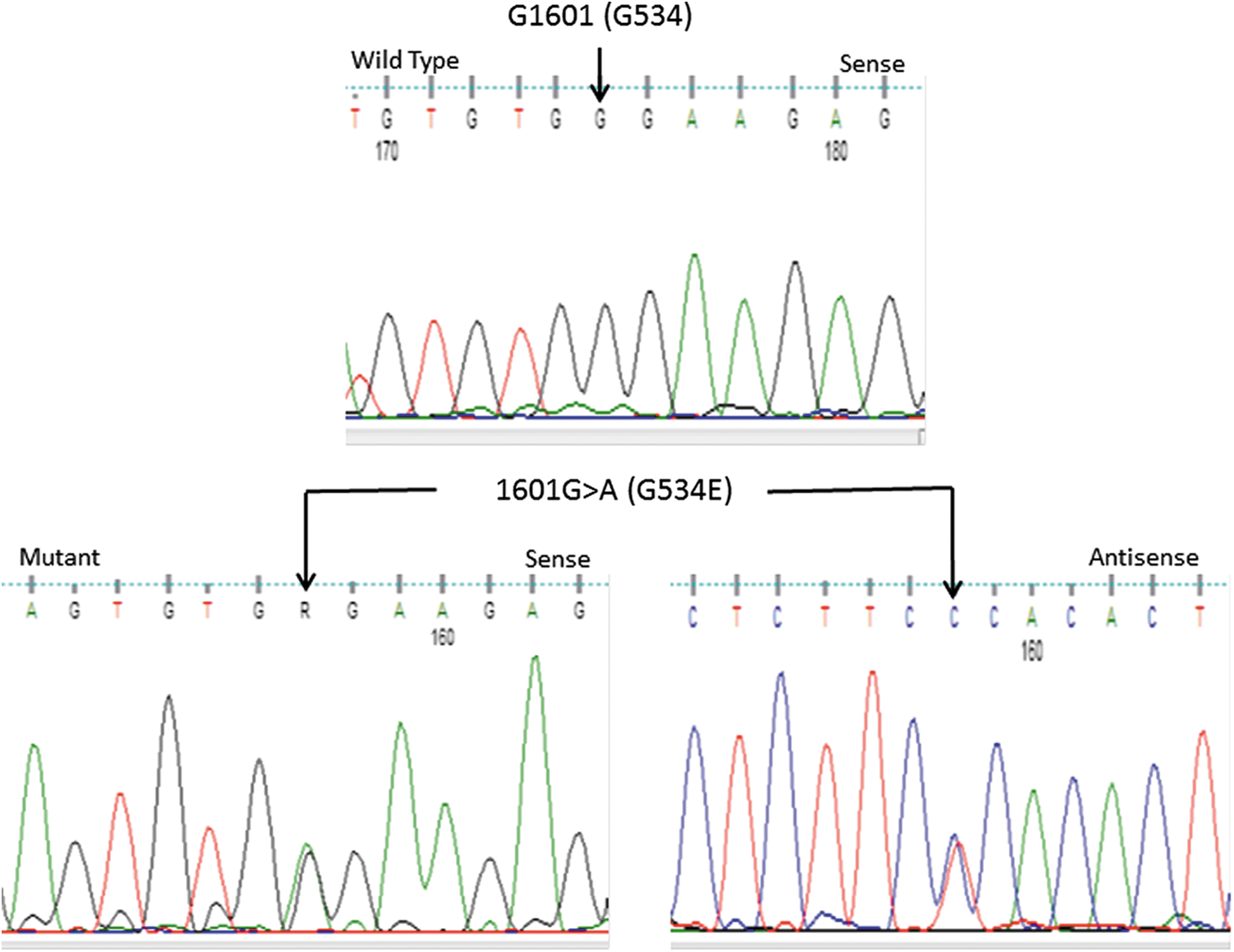
Sequence chromatograms showing a normal sequence of a segment of exon 13 of the HABP2 gene (upper panel) and a heterozygous 1601G>A mutation in sense and antisense directions (lower panel) in a patient with sporadic papillary thyroid cancer. This nucleotide change leads to replacement of glycine by glutamate at position 534 (G534E). Color images available online at
HABP2 G534E mutation in a random sample of NMTC using peripheral leucocyte DNA
None of 217 cases of NMTC screened for the G534E HABP2 mutation using genomic DNA isolated from peripheral leucocytes harbored this mutation.
HABP2 G534E mutation in normal control sample
Of 190 normal control subjects, only one carried a monoallelic HABP2 G534E mutation. This was a 27-year-old man without any thyroid illness and no goiter on physical examination. His family history was completely negative for thyroid diseases.
Overall results of HABP2 mutation screen
In a sample of 520 cases of familial (11 cases), pediatric (63 cases), and adult (446 cases) NMTC, only one case (0.2%) harbored the G534E mutation. In a sample of 190 normal subjects, one individual (0.53%) also harbored the G534E mutation.
Discussion
The recently published work by Gara et al. on the role of the HABP2 G534E mutation as a possible underlying cause of PTC in a seven-member family with familial NMTC was an exciting finding that was hoped to explain the development of NMTC in a significant number of cases with familial NMTC (6). The authors also reported that this mutation was found in 4.7% of 423 apparently sporadic cases of TC (6). We were particularly interested in these findings, since TC is very common in our population, being the second most common malignancy in females and the sixth most common cancer in the population at large (15). Familial and pediatric cases of TC are also common (16). In addition, our society has a high rate of consanguineous marriage and hereditary diseases (13,14). All of these factors made it logical to predict a high rate of this mutation in our patients. Surprisingly, only two subjects carrying these genetic variants were found: one with a sporadic NMTC, and one normal control subject. It was not found in a large sample of TC patients, including 11 cases of four families with NMTC (Table 1), 63 cases of pediatric TC, 446 adult cases of NMTC, and 190 normal controls. This strongly suggests that this variant is very rare and does not play a role in the pathogenesis of TC in this population. In the familial forms of NMTC, no mutations in the all exons and splicing sites of the HABP2 gene were found. The reason for these somewhat surprising findings is not fully clear. It is possible that this is due to racial/ethnic differences, but it could also suggest heterogeneity of familial NMTC. A recent review suggested that the genetics of NMTC are complex and are likely to be related to several genetic loci with mild to moderate effects (5). It also suggested that the predisposing genes are likely to be non-coding regulatory in nature, and this may lead to missing these causative genes on whole exome sequencing (5). Although the work by Gara et al. has several lines of evidence suggesting that the HABP2 was the underlying genetic defect in the family reported, some questions remain, including the fact that HABP2 was only reported in a single family and was not confirmed in other families and there was no in vivo work to support the in vitro findings. Unlike the usual two-hit mechanism by which tumor suppressor genes cause cancer, the G534E variant reported in this kindred was heterozygous in both the tumor tissue and the peripheral blood. However, the authors suggested that the HABP2 is a tumor suppressor gene with a dominant negative effect. Finally, the G534E variant is a rare known single nucleotide polymorphism (SNP) that is present in the normal population (6).
Familial NMTC can be part of well-recognized hereditary syndromes in which NMTC is a component (5,10). These syndromes were mentioned above and are mostly known to be due to defined genetic mutations. Apart from these rare well-known syndromes that are associated with familial NMTC, the existence of non-syndromic familial NMTC as a distinct entity had been questioned for a long time (4,17 –21). Although it is not uncommon to encounter TC in more than one family member, TC is common, and this may reflect a chance co-occurrence. In addition, it may reflect environmental factors with or without genetic predisposition. It has been suggested that about 62–69% of patients with another affected relative are suffering from a sporadic rather than a familial form of NMTC (22). This percentage also depends on how familial NMTC is defined. If two affected members of the family are considered an adequate number to define familial NMTC, there is a chance for over-diagnosing this condition (23). The specificity of the definition improves significantly if more than two family members are required for the diagnosis of familial NMTC (5). However, several lines of evidence suggest that familial NMTC is a distinct entity.
Large case-control and cohort studies found that patients with familial NMTC are generally younger than the sporadic forms of NMTC, suggesting the existence of a strong predisposing factor that is likely to be genetic in nature (23). Familial NMTC has been linked to a number of genetic loci through linkage analysis of large families with a number of affected members. In one family with two members affected by PTC and 18 members having multinodular goiters, linkage analysis with all 20 members considered to be affected strongly suggested that 14q32 as a likely contributory locus. This locus was named multinodular goiter 1 (MNG1) (24). Although it was significantly associated with MNG and PTC in this family (LOD score 3.8), it was not associated with NMTC in 37 smaller pedigrees each containing at least two cases of NMTC (24). A large multicenter study including 191 members of 80 families from different parts of the world showed linkage to 2q21 locus with a LOD score 3.07 (25). In a large Tasmanian pedigree reported in the same study, this locus was found in 7/8 members affected by PTC (25). Furthermore, in a subset of 10 families from the same study, linkage was shown for the 19p as well as the 2q21 loci (25). Other linkage studies identified loci at 8.p23.1–p22, 19p13.2, 1q21, and 6q22 (reviewed in Nagy and Ringel) (5). More recent studies used genome-wide analyses to look for SNPs that may be associated with familial NMTC (5). These studies identified a large number of SNPs with variable odds ratios (5). However, no single gene was characterized as a definite underlying susceptibility gene in familial NMTC at a large scale.
It was hoped that the HABP2 G534E variant could be a common gene defect in a significant percentage of patients with familial NMTC. The present study does not support this, but other studies from other racial/ethnic groups need to be performed before such a conclusion can be made more definitively. Although the number of patients included in this study is large, familial cases remained relatively small, and it is possible that HABP2 mutations might be the underlying genetic defect in other families with familial NMTC. However, a relatively large sample of pediatric NMTC was included, a group of patients that could have a strong genetic predisposition. In addition, a large sample of adult NMTC included in the analysis did not harbor the HABP2 G534E mutation, although 4.7% of 423 adult patients reported by Gara et al. were found to carry the G534E allele (6). Except in the familial cases of NMTC, the other exons of the HABP2 gene were not screened for mutations other than G534E mutation in the coding region. However, such mutations were not found in the familial cases of NMTC where one would expect such mutations to occur. Similarly, Gara et al. did not find other mutations in the HABP2 gene in the family that they described (6).
In summary, this study could not confirm that HABP2 is a susceptibility gene in a large sample of familial and sporadic NMTC in a highly inbred population in the Middle East.
Footnotes
Author Disclosure Statement
The authors have nothing to disclose.