
Abstract
Background:
Congenital hypothyroidism of central origin (CH-C) is a rare disease in which thyroid hormone deficiency is caused by insufficient thyrotropin stimulation of a normal thyroid gland. A recently described syndrome of isolated CH-C and macroorchidism was attributed to loss-of-function mutations of the immunoglobulin superfamily, member 1 gene (IGSF1).
Patients and Methods:
CH-C was diagnosed in three siblings. The TRH, TRHR, and TSHB genes were sequenced followed by whole-exome sequencing in the proband. A mutation identified in IGSF1 was analyzed by direct PCR sequencing in family members. The effects of the mutation were assessed by in vitro studies in HEK293 cells.
Results:
The index case was negative for mutations in TRH, TRHR, and TSHB. Whole-exome sequencing revealed a novel insertion mutation in IGSF1, c.2284_2285insA, p.R762QfsX7, which was confirmed by direct PCR sequencing and was identified in six additional family members. The mutation introduces a frame-shift and premature stop codon in the seventh Ig loop, thereby truncating IGSF1. In vitro studies revealed that the mutated IGSF1–R762QfsX7 migrates as a doublet at ∼28 kDa, which is far smaller than the wild type protein (130–140 kDa). Both bands were endonuclease H sensitive, indicating immature glycosylation and failure of the protein to traffic out of the endoplasmic reticulum to the plasma membrane. Further phenotypic findings in the family included macroorchidism and infertility in the uncle and mild neurological phenotypes in the affected males, such as hypotonia, delayed psychomotor development, clumsy behavior, and attention deficit disorder.
Conclusions:
We identified a novel insertion mutation in the IGSF1 gene and further delineated the phenotype of the IGSF1-deficiency syndrome. Our findings indicate a possible association between an IGSF1 mutation and neurological phenotypes.
Introduction
C
Loss-of-function mutations of the immunoglobulin (Ig) superfamily, member 1 (IGSF1; OMIM 300888) were recently described and linked to a novel syndrome of isolated CH-C and macroorchidism (10). The IGSF1 gene is located on the X chromosome (Xq26.1) and encodes a plasma membrane glycoprotein that is mainly expressed in the pituitary, brain, and testis (11). The IGSF1 protein has 12 Ig-like loops of the C2 (constant region type 2) type, a transmembrane domain and a short intracellular cytosolic tail. Only the C-terminal domain of the protein, containing seven Ig loops, traffics to the plasma membrane (12,13). Many members of the Ig superfamily are cell-adhesion molecules that regulate signaling via cell-surface receptors and are involved in the immune and hematological systems as well as the development of the central nervous system and adult neural cell functions (14). The exact roles of IGSF1 in pituitary hormone secretion and testicular function remain undefined.
The main characteristics of the IGSF1 deficiency syndrome include central hypothyroidism, macroorchidism, delayed pubertal testosterone secretion, increased body mass index (BMI) and variable prolactin and growth hormone (GH) deficiencies (15,16). Sun et al. (10) described 26 patients with 10 distinct IGSF1 mutations (10). Since that first report, an additional five mutations have been described in Japanese patients (17 –19) and in one male patient carrying a Xq26 deletion encompassing the IGSF1 gene (20).
Here, we describe a family with three siblings diagnosed with isolated CH-C. Using whole-exome sequencing (WES), we identified a novel insertion mutation in the IGSF1 gene, c.2284_2285insA, p.R762QfsX7 in the index case, in his two brothers and in four family members. We further delineate the phenotype of this rare condition, expressed the mutant protein in vitro, and correlated genotype and phenotype in the family.
Patients and Methods
Patients
Blood samples for hormonal and DNA analyses were collected from all participants. Medical history and laboratory data were collected from the family members' computerized medical files. The TRH, TRHR, and TSHB coding regions were sequenced (see below). Because no point mutations were found in these genes, we further performed WES on the index case (patient III-1). After identification of a novel IGSF1 variant, we recruited eight family members and analyzed the identified IGSF1 gene mutation by direct sequencing of PCR amplicons. The study was approved by the Ha'Emek Medical Center Ethics Committee and the National Helsinki Committee for Genetic Studies. Informed consent was obtained from the study participants.
Israeli national neonatal screening for CH
Blood samples are routinely collected by heel puncture 48–72 hours after birth. Neonatal thyroxine (T4) function is measured by PerkinElmer B065-112 AutoDELFIA total T4 (TT4) and B032-312 AutoDELFIA (TSH), both using time-resolved fluoroimmunoassays. The Israeli screening program is based on a TT4 test followed by a confirmatory TSH test. Therefore, when the level of TT4 is below the daily 10th percentile, TSH is measured. TSH values above 20 mIU/L are considered indicative of primary CH. Neonates with abnormal screening results are referred to medical centers.
Sequencing of TRH, TRHR, and TSHB
Genomic DNA was isolated from blood leukocytes using MasterPure™ DNA Purification Kit for Blood Version II from Epicenter (Illumina) according to the manufacturer's instructions. The coding sequences of TSHB (exons 2 and 3), TRH (exons 1 to 3), and TRHR (exons 1 and 2) were amplified by PCR using 20 ng genomic DNA as the template supplemented with 1 U Taq polymerase (Axon Labortechnik), 1 × PCR buffer and 1.5 mM magnesium chloride (Axon Labortechnick), 10 μM dNTPs (Promega Corporation), 1 M betaine (Sigma-Aldrich Inc.), and 0.4 μM each of forward and reverse primers (Supplementary Table S1; Supplementary Data are available online at
Whole-exome sequencing
Genomic DNA was extracted from peripheral blood mononuclear cells. WES was used to search for candidate variants in the index case (in a recessive model). Genomic DNA libraries were created following standard protocols. Libraries were enriched using a Nextera Exome enrichment kit (62 Mbp; Illumina) and sequenced on a HiSeq 2500 sequencer (2 × 170 bp pair ends run) at an 83 × mean coverage. Reads were aligned to the human genome (hg19; National Center for Biotechnology Information build 37; February 2009). Exome-filtering criteria for both families are summarized in Supplementary Table S2. Sanger sequencing was used to validate all coding and splicing variants in the family that were homo/hemizygous and had a positive logarithm (base 10) of odds score, and were absent or found at very low frequencies in dbSNP, Exome Variant Server (
Sequence analysis of IGSF1
Exon and exon–intron boundaries of IGSF1 were amplified from genomic DNA by PCR using previously reported primers (17). PCR products were sequenced directly with an ABI 373A automated fluorescent sequencer (PE Applied Biosystems). Sanger sequencing was performed for the identified mutation in all subjects. PCR products were sequenced using BigDye Terminator v.3.1. Sequence analysis was performed on an Applied Biosystems 3130xl Genetic Analyzer.
In vitro analysis of mutant IGSF1 protein
Human embryonic kidney (HEK) 293 cells were cultured as described by Sun et al. (10). QuikChange mutagenesis was performed using primers (Forward: 5′-CTC ACA CTG AAA AAA CGC CCC TTC AAG TG-3′; Reverse: 5′-CAC TTG AAG GGG CGT TTT TTC AGT GTG AG-3′) to introduce the 2284_2285insA mutation into the previously described wild type IGSF1-HA expression vector (12). Cells were seeded in six-well plates and transfected with 2 μg of plasmid DNA using 6 μg polyethylenimine. Total protein lysates were prepared 24 hours post-transfection using immunoprecipitation lysis buffer (50 mM Tris pH 7.5, 150 mM sodium chloride, 1 mM ethylenediaminetetraacetic acid, 1% v/v Triton X-100). Peptide-N-Glycosidase F and endonuclease H (EndoH) treatments as well as protein Western blotting were performed as described previously (10).
Results
Patient reports
Index case (patient III-1)
The index case was a two-week-old male born to unrelated parents of Ashkenazi Jewish decent. Following a prenatal ultrasound scan that showed a short femur, amniocentesis revealed a normal male karyotype 46, XY. He was born by spontaneous delivery at 38 weeks gestational age with a weight of 2770 g. After birth, bradycardia was observed but cardiac echography demonstrated a normal heart. At the age of 2 weeks, he was referred to our hospital due to prolonged neonatal jaundice (20 mg/dL bilirubin) with elevated liver enzymes (aspartate aminotransferase 104 U/L, alanine aminotransferase 64 U/L, lactic dehydrogenase 965 U/L, alkaline phosphatase 965 U/L). As part of the investigation of the prolonged neonatal jaundice, thyroid functions were checked, revealing a low free T4 (FT4) of 0.47 ng/dL (normal infant range, 0.83–2.0 ng/dL) with a normal TSH of 2.9 mIU/L (normal infant range, 0.5–10 mIU/L) (Table 1). The results of the neonatal screening after the initial diagnosis revealed low TT4 with low TSH (Table 1), consistent with the diagnosis of CH-C. On examination, he was severely hypotonic with severe head lag. Levothyroxine (
Dashes indicate no value for the patient.
Adult normal values.
The Israeli screening program is total T4-based; when the level of total T4 is below the daily 10th percentile, TSH is measured.
Normal FT4 levels for infant aged 1–30 days, 0.83–2.0 ng/dL.
Normal TSH levels for infant aged 1–30 days, 0.5–10 mIU/L.
NA, not available; ND, not done; T3, triiodothyronine; T4, thyroxine; TSH, thyrotropin; LH, Luteinizing hormone; 99TC, technetium-99m; IGF, insulin-like growth factor; SD, standard deviation.
Patient III-3 (brother of the proband)
Patient III-3 was an infant male born following a normal pregnancy, weighing 3160 g at term. Thyroid function tests were performed at the age of 7 days due to his brother's CH and revealed low FT4 with normal TSH and FT3 levels (Table 1). The results of the neonatal screening that were reported after his first visit revealed a borderline low TT4 with a normal TSH (Table 1). On examination, he had severe head lag without any other findings.
Patient III-5 (brother of the proband)
Patient III-5 was a 3-day-old male born following a pregnancy with polyhydramnios, weighing 4030 g at 42 weeks gestational age. Neonatal screening had shown normal TT4 and TSH levels (Table 1), and thyroid function at the age of 2 days was also within the reference range for his age (TSH = 15.7 mIU/L, FT4 = 0.84 ng/dL). Repeated thyroid function tests at the age of 7 days were also within the reference range (TSH = 3.61 mIU/L, FT4 = 0.99 ng/dL, and FT3 = 3.3 pg/mL). On examination, he had severe head lag and neonatal jaundice without other signs of hypothyroidism. At the age of 6 weeks, repeated thyroid function tests revealed a borderline low FT4 with normal serum levels of TSH and FT3 (Table 1); he was diagnosed with CH-C and
Patient II-2 (uncle of the index case)
A 27-year-old male was evaluated at our clinic due to his nephew's familial CH. After birth, he had been diagnosed by the national neonatal screening with CH (data not available); however, he never received supplemental
Family history
The mother and father were both healthy and had normal thyroid function tests (Fig. 1). Parental height was 183 cm and maternal height 164 cm. In addition to the three affected sons, they had two daughters with normal psychomotor development and normal thyroid function. The maternal grandmother had had a toxic multinodular goiter and had been treated with radioactive iodine, resulting in acquired hypothyroidism. Another uncle (II-1) was diagnosed with hypothyroidism at the age of three months, but he was never treated. He had significant psychomotor delay at infancy and as an adult he had clumsy motor behavior, as well as speech problems. He declined to participate in the study.

Pedigree of the Israeli family with the novel IGSF1 gene mutation. The age (in years) indicates the age of each subject at the time of analysis. In patients III-1, III-3, and III-5, the data represent thyroid function prior to levothyroxine (
Genetic results
The index case was negative for mutations in the TRH, TRHR, and TSHB genes. Using WES, we identified a total of 25,100 variants, which were filtered according to various criteria (Supplementary Table S2). From the eight remaining variants, only the IGSF1 mutation segregated with the abnormal phenotype in the family. Sanger sequencing confirmed the presence of the mutation. A novel mutation, c.2284_2285insA, p.R762QfsX7, was identified in IGSF1 in the proband (Fig. 2). IGSF1 is located on the X chromosome and the mutation was hemizygous in the three affected siblings and the maternal uncle and heterozygous in the mother, one sister, and the maternal grandmother (Fig. 1).
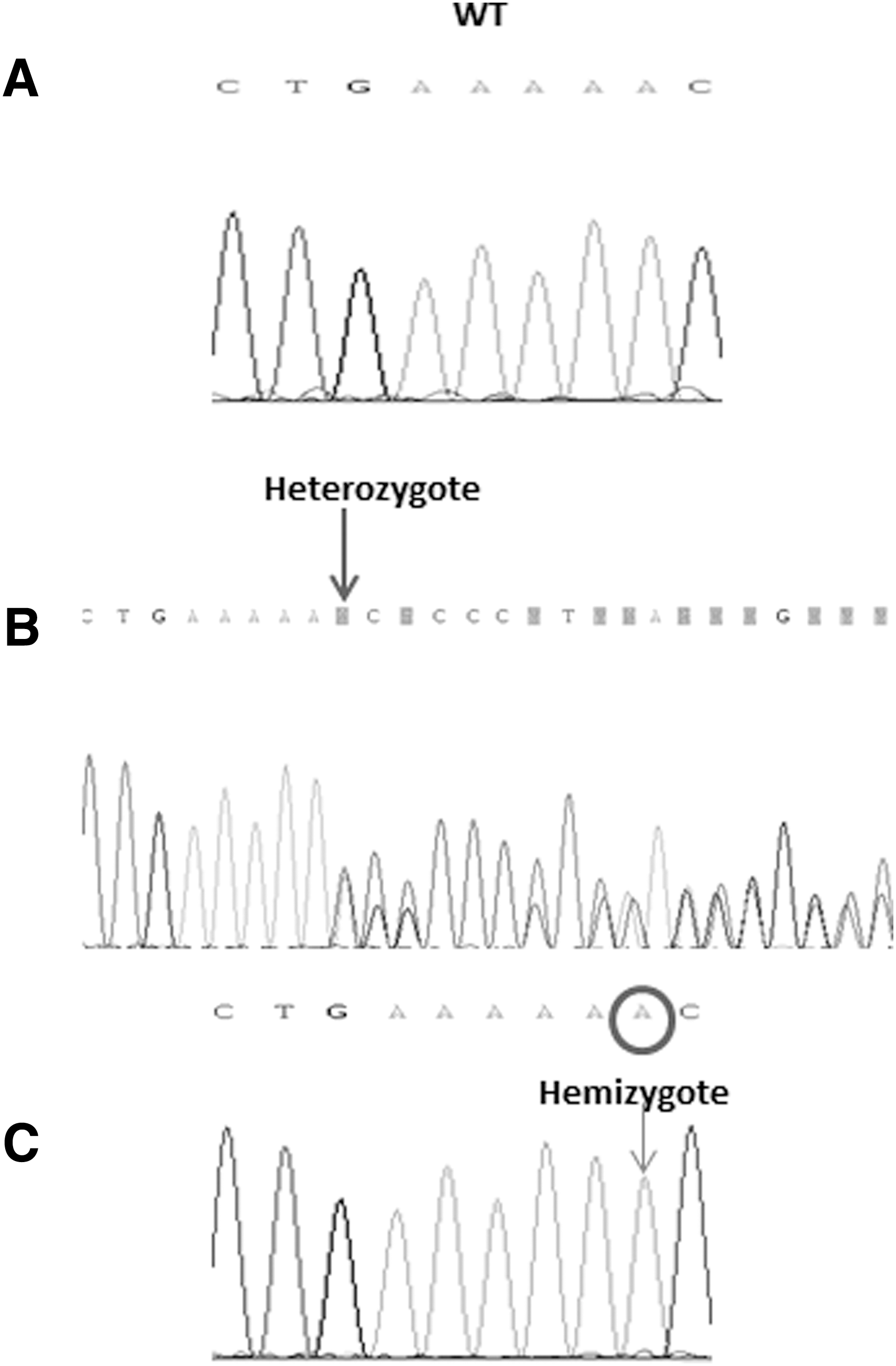
Sequence of the IGSF1 gene in (
We next assessed the pathogenicity of the identified mutation by expressing the p.R762QfsX7 mutant in heterologous HEK293 cells. The wild type IGSF1 migrates as a doublet on an sodium dodecyl sulfate–polyacrylamide gel (Fig. 3A, lane 2). As previously reported (10), the higher molecular weight band reflects the mature (EndoH-resistant, lane 3) glycoform of the protein that traffics to the plasma membrane, whereas the lower molecular weight band represents the immature (EndoH-sensitive) glycoform that is retained in the endoplasmic reticulum (ER) (10). The identified novel mutation introduces a frame shift and a premature stop codon in the seventh Ig loop (the second Ig loop of the C-terminal domain), truncating the IGSF1 protein. When examined by Western blot, IGSF1–p.R762QfsX7 migrated as a low abundance doublet at ∼28 kDa (Fig. 3B, lane 2), which is far smaller than the wild type protein at ∼130–140 kDa. In contrast to the wild type, both bands of the IGSF1–p.R762QfsX7 doublet were EndoH sensitive (Fig. 3B, lane 3), indicating immature glycosylation and failure of the protein to traffic out of the ER to the plasma membrane. Similar results have been reported with other truncation mutants (10). The generated protein is small and unstable, with impaired transport out of the ER.
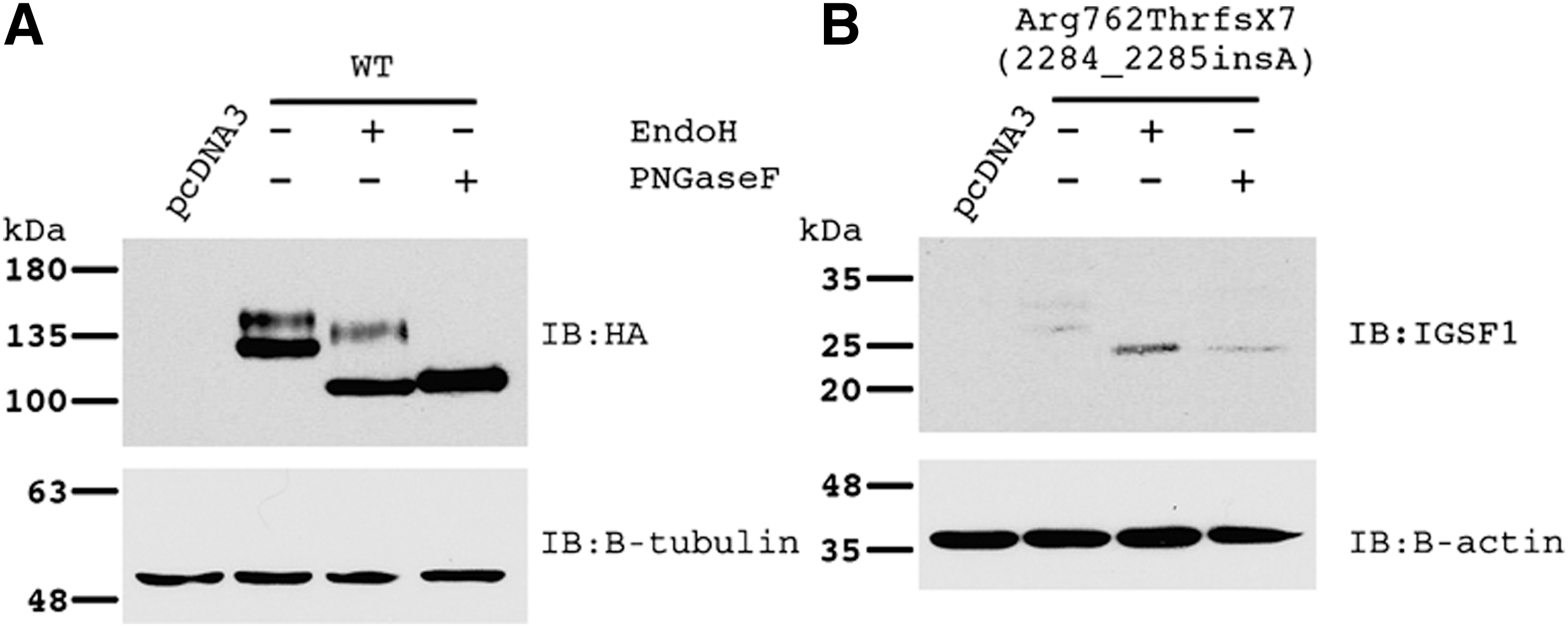
Protein analysis of wild type (
Discussion
In three siblings with CH-C and four other family members, we identified a novel insertion mutation, c.2284_2285insA, p.R762QfsX7, which introduces a frame shift and thus a premature stop codon in the seventh Ig loop of IGSF1. Sun et al. (10) identified 10 distinct mutations in the IGSF1 gene in males from 11 unrelated European families presenting with central hypothyroidism and testicular enlargement. An additional five novel IGSF1 mutations were described in Japanese patients presenting with either CH-C or failure to thrive (17
–19). In all, 15 different mutations have been reported to date in IGSF1, including 6 nonsense, 5 missense, 1 insertion, and 3 deletion mutations. The main initially described phenotypic characteristics of patients with IGSF1 deficiency include central hypothyroidism and macroorchidism, but further clinical characterization of these patients also revealed hypoprolactinemia, variable GH deficiency, delayed pubertal testosterone secretion with normal timing of testicular development and increased body weight (16). Since IGSF1 is located on the X chromosome, the affected patients are males, but a subset of female carriers also exhibit mild central hypothyroidism (16). In the family reported here, all affected males had CH-C but only three were treated with
The exact frequency of IGSF1 mutations is not known, but a frequency of approximately 1:100,000 has been predicted (15). Adult male Igsf1-deficient mice exhibit reduced pituitary and serum TSH and reduced serum FT3 and/or FT4 concentrations, likely due to impaired TRH signaling (10). The clinical phenotype of IGSF1 deficiency in humans with respect to thyroid function is similar to that of mice lacking Igsf1.
CH-C is only detected by TT4-based neonatal screening and is missed by TSH-based screening. Since the Israeli national newborn screening program for CH is based on a TT4 test followed by a confirmatory TSH test, these cases are identified by the screening program, whereas in TSH-based screening they are missed and the diagnosis is made later in infancy or childhood following evaluation of prolonged neonatal jaundice, failure to thrive, short stature, or delayed psychomotor development. Furthermore, some patients are diagnosed with CH-C only after familial genetic analysis in the fifth to eighth decade with no clinical symptoms of hypothyroidism (10,16). Interestingly, while patients III-1 and III-3 were identified by the national screening program, in the third sibling (III-5) the TT4 screening levels were within the 10th percentile of the normal range. Only at the age of six weeks did laboratory FT4 levels decrease below the reference range, indicating that the hypothyroidism developed over time. These findings demonstrate variation in the extent of hypothyroidism within a family. Moreover, despite the fact that he was never treated with supplemental
IGSF1 has 12 Ig-like C2-type loops, a transmembrane domain, and a short intracellular cytosolic tail. The protein is cleaved into N- and C-terminal domains and only the latter traffics to the plasma membrane (12,13). Using in vitro studies in HEK293 cells, we show that the identified p.R762QfsX7 mutation causes a premature truncation of the IGSF1 C-terminal domain, which does not acquire complex carbohydrates. This indicates that the protein does not traffic from the ER through the Golgi to the plasma membrane. Similar results have been reported with other IGSF1 truncation mutations (10,17,18). Indeed, all of the intragenic mutations identified to date, apart from one (19), have been located in the C-terminal domain, indicating its important role in IGSF1 function.
IGSF1 is expressed in rodent pituitary thyrotrophs, lactotrophs, and somatotrophs (10,11). Consistent with this pattern of expression, the phenotype of IGSF1 deficiency in humans includes central hypothyroidism and variable prolactin and GH deficiencies (16). In our family, all affected males had normal growth patterns and the adult male was within the normal range for adult height, indicating a lesser prominence of GH deficiency in this syndrome. Hypoprolactinemia has been reported in about 70% of male patients (10,16) and in two of our patients, but its clinical significance in males is still undefined. Mice lacking Igsf1 are fertile and have normal testicular volumes (10). IGSF1-deficient males exhibit macroorchidism but are fertile and have normal gonadotropin and testosterone levels (10,16). Pubertal development has been reported to be disharmonious, with testicular growth starting at a normal age, but testosterone secretion, pubertal spurt and the development of pubic hair being delayed (16). It is not clear when testicular enlargement develops; however, enlarged testes have been observed at as early as 11 years of age. In the family described here, all three young (prepubertal) siblings had normal testicular volumes, whereas patient II-2, who was 27 years old, exhibited macroorchidism, abnormal sperm motility and increased pathological morphology. Since only one adult male exhibited infertility in this family, other etiologies for abnormal spermatogenesis cannot be excluded.
It was initially hypothesized that IGSF1 acts as an inhibin co-receptor, and the consequence of loss of inhibin action was expected to result in enhanced follicle-stimulating hormone (FSH) secretion; however, further studies challenged this hypothesis (22). The association between hypothyroidism and macroorchidism has been reported in the past in males with prolonged untreated hypothyroidism (23 –25). It has been hypothesized that macroorchidism results from increased FSH stimulated by hypothalamic TRH, which in turn stimulates the proliferation of Sertoli cells (which determines testis size) (25). Interestingly, the increase in basal and pulsatile 24 hour FSH in IGSF1 deficient males compared with controls might at least partially explain the macroorchidism (26). Furthermore, T3 itself has direct effects on Sertoli cell proliferation (27,28). Our finding is supported by a study showing that IGSF1 is expressed in elongating spermatids in rats (11), and by the fact that the other members of the Ig superfamily are expressed at the membrane of germ cells and spermatozoa, where they play roles in cell adhesion and sperm–egg interactions (11). In other reports of adult males with macroorchidism, sperm analysis was not reported.
Our findings of hypotonia, delayed motor development, ADD, and clumsiness (without cognitive impairment) in the IGSF1-deficient males may point to a role for IGSF1 in neural cell functions. Indeed, the neural cell adhesion molecules in the Ig superfamily have previously been implicated in the development of the central nervous system, facilitating neural cell migration, axon guidance, and synapse formation (14). In adult life, they maintain synaptic connections, cell–cell contacts, neuron–glial interactions, and synaptic plasticity (14). Dysfunction of these cells has been linked to psychiatric conditions as well as to Alzheimer's disease and autism (14). We have shown an association between mutations in IGSF1 and subtle neurological symptoms in affected males. It might be assumed that this subset of neurological symptoms is due to delayed initiation of
In conclusion, we identified a novel mutation in IGSF1 and further delineate the phenotype of the IGSF1-deficiency syndrome. The data presented here suggest an association between IGSF1 mutations and neurological phenotypes.
Footnotes
Acknowledgments
We thank Camille Vainstein for professional language editing and the families for participating in and providing samples for this study. This work was supported by a grant from the Rashut Lepituach HaGalil and the Negev (Y.T.-R.) and by operating grants from the Canadian Institutes of Health Research (MOP-133557) and Natural Sciences and Engineering Research Council of Canada (2015-05178) to D.J.B. M.-O.T. was supported by fellowships from Réseau Québécois en reproduction and Natural Sciences and Engineering Research Council of Canada.
Author Disclosure Statement
The authors have nothing to disclose.