
Abstract
Background:
Hemizygous mutations in the immunoglobulin superfamily member 1 (IGSF1) gene have been demonstrated to cause congenital central hypothyroidism in males. This study reports a family with a novel mutation in the IGSF1 gene located on the long arm of the X chromosome.
Patient findings:
A two-month-old boy was diagnosed with central hypothyroidism because of prolonged jaundice. A thyrotropin-releasing hormone (TRH) stimulation test indicated dysfunction in both the hypothalamus and the pituitary gland, and prompted the IGSF1 gene to be analyzed. The patient had a novel nonsense variant, c.2713C>T (p.Q905X), in exon 14 of the IGSF1 gene. Studies of the family revealed that the patient's sister and mother were heterozygous carriers of the IGSF1 mutation. The patient's maternal uncle carried the same mutation as the proband but had no overt symptoms. The mother and uncle started levothyroxine supplementation because of subclinical hypothyroidism.
Summary:
A novel mutation (c.2713C>T, p.Q905X) of the IGSF1 gene was identified that causes congenital central hypothyroidism in a Japanese family. The findings further expand the clinical heterogeneity of this entity.
Introduction
C
The pathogenic mechanisms of CCH are heterogeneous, and dysfunction of thyrotroph-specific genes, such as the thyroid-stimulating hormone β-subunit (TSHβ) and the TRH receptor (TRHR) can result in isolated central hypothyroidism (5,6). Many CCH patients, however, have additional pituitary hormone deficiencies (7). Some patients with combined pituitary hormone deficiencies (CPHD) were reported to carry gene mutations of transcription factors involved in pituitary development, including POU1F, PROP1, HESX1, and LHX3 (8).
Sun et al. (9) recently reported X-linked central hypothyroidism caused by a loss-of-function mutation in the immunoglobulin superfamily member 1 (IGSF1) gene. IGSF1, located on Xq26.2, encodes a plasma membrane immunoglobulin superfamily glycoprotein (10). After proteolytic cleavage, the C-terminal portion traffics to the plasma membrane where it is expressed as a large extracellular domain, suggesting a possible function in cell–cell adhesion or signaling (11). IGSF1 is expressed in Rathke's pouch, the adult pituitary gland, and the hypothalamus (9,12)
In addition to CCH, the IGSF1 deficiency syndrome is characterized by postpubertal macro-orchidism, preceded by disharmonious puberty, hypoprolactinemia, and transient partial growth hormone deficiency (GHD) in childhood (13).
Here, a Japanese family is reported with IGSF1 deficiency associated with a novel mutation in the IGSF1 gene.
Patients
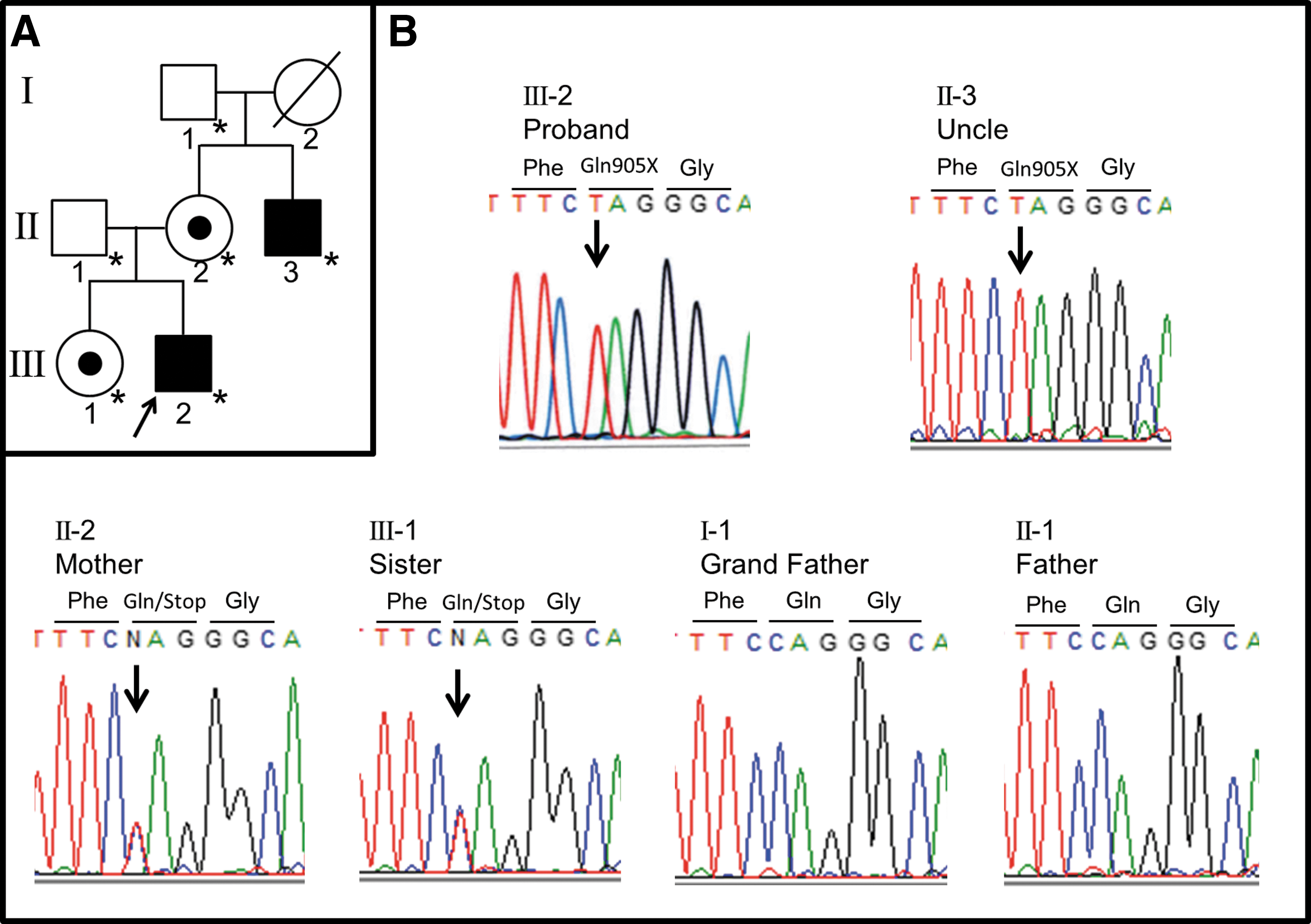
The pedigree of the proband and his family is shown in Figure 1A. The proband is denoted by the arrow. This study was approved by the Institutional Review Board of Osaka City University Graduate School of Medicine, Osaka, Japan (#3375). After obtaining written informed consent from the patient's family for the molecular study, genomic DNA was extracted from peripheral blood leukocytes. Exon and exon–intron boundaries of the IGSF1 gene were amplified by polymerase chain reaction (PCR) using primers as reported previously (9). After amplification, PCR products were purified and sequenced directly with an ABI PRISM Dye Terminator Cycle Kit and an ABI 373A automated fluorescent sequencer (PE Applied Biosystems, Foster City, CA). The genetic analysis revealed a novel hemizygous IGSF1 mutation (c.2713C>T, p.Q905X) in the patient (Fig. 1B). After obtaining written informed consent, mutational analysis was also performed in his parents, older sister, maternal uncle, and maternal grandfather (his maternal grandmother was deceased). The family analysis revealed that the proband's mother and sister are heterozygous carriers of the mutation. The maternal uncle had the same mutation as the proband in a hemizygous state. The grandfather did not carry the mutation (Fig. 1B).
(
Patient Report
Patient 1 (the proband: III-2)
The patient, a male, was the second child of healthy unrelated parents. The family history was unremarkable. He was born by normal vaginal delivery at the 40th week of gestation. The birth weight was 3820 g (+1.4 SD), length was 51.2 cm (+0.8 SD), and head circumference was 34.8 cm (+1.0 SD). The Apgar scores were 8 and 9 at one and five minutes, respectively. Neonatal screening for hypothyroidism based on thyrotropin (TSH) measurement did not detect any abnormality.
At the regular one-month checkup, prolonged neonatal jaundice was noted (total bilirubin 19.3 mg/dL [reference range 0.25–2.3 mg/dL]; direct bilirubin 1.1 mg/dL [reference range 0.2–0.8 mg/dL]). As thyroid function tests revealed central hypothyroidism (TSH = 3.26 mIU/L [reference range 0.5–6.5 mIU/L]) and free thyroxine [fT4] = 0.49 ng/dL [reference range 0.8–2.1 ng/dL]), the patient was referred to hospital for further examination.
On admission, he was two months old, his weight was 6775 g (+0.3 SD), length was 58.6 cm, and body mass index (BMI) was 19.7 kg/m2 (+2.3 SD). The patient did not present any dysmorphic features or goiter, and his bilateral testicular size was 1 mL measured with an orchidometer. He was jaundiced, but his physical exam was otherwise unremarkable. Radiography detected bilateral distal femoral epiphyses. Laboratory findings are summarized in Table 1. His total bilirubin level (14.3 mg/dL) was increased. Thyroid function tests showed a decreased level of fT4 with an inappropriately normal TSH level in the serum (Table 1). A combined pituitary function test was performed. A thyrotropin-releasing hormone (TRH) stimulation induced a prompt but low-level TSH response, indicating pituitary dysfunction. The TSH response was prolonged after the peak, and the GH response to arginine was delayed, suggesting hypothalamic involvement (Fig. 2). Serum basal and stimulated PRL levels after TRH stimulation were normal. The basal serum cortisol level was within the reference range. Magnetic resonance imaging (MRI) of the brain showed a normal hypothalamic–pituitary region. Based on these findings, he was diagnosed as having isolated CCH, and thus levothyroxine therapy was started. Thereafter, his jaundice improved. The patient is currently 17 months old, and his growth and developmental milestones are within the reference range.
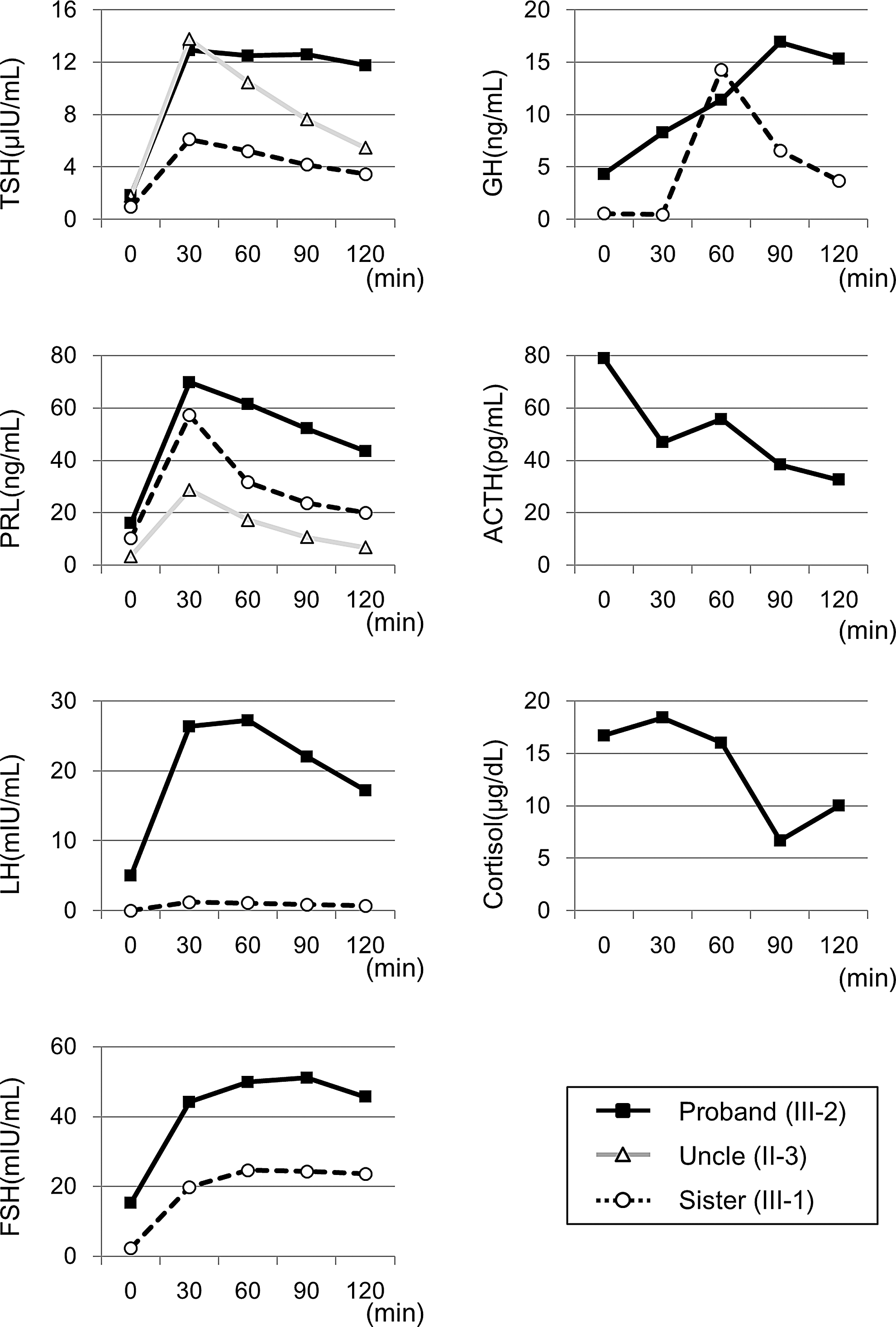
The results of the thyrotropin-releasing hormone, gonadotropin-releasing hormone, arginine, and corticotrophin-releasing hormone stimulation tests.
All blood samples were collected during a fasting state in the morning.
The proband and his uncle were hemizygous for the mutation.
The mother and sister were heterozygous for the mutation.
Normal range for this age: TSH = 0.7–6.5 μIU/mL; fT3 = 2.3–4.6 pg/mL; fT4 = 1.0–1.9 ng/dL.
Normal range for this age: TSH = 0.3–3.9 μIU/mL; fT3 = 2.3–4.4 pg/mL; fT4 = 1.0–2.0 ng/dL.
Normal range for 2-year-old boy: 18–154 ng/mL; 4-year-old girl: 48–238 ng/mL; 36-year-old man: 99–275 ng/mL; 38-year-old woman: 103–254 ng/mL.
TSH, thyrotropin; fT3, free triiodothyronine; fT4, free thyroxine; PRL, prolactin; LH, luteinizing hormone; FSH, follicle-stimulating hormone; ACTH, adrenocorticotropic hormone; COR, cortisol; T, testosterone; TG, triglyceride; HDL, high-density lipoprotein cholesterol; LDL, low-density lipoprotein cholesterol; ND, not determined.
Patient 2 (maternal uncle of the proband: II-3)
He was 36 years old at the time of the study. His height was 168.8 cm, weight was 73.8 kg, BMI was 25.9 kg/m2, and abdominal circumference was 86 cm. His testicular volumes were 25 mL bilaterally. He has been healthy, although he had a delayed pubertal growth spurt by history. He graduated from a regular high school, and was working as a full-time employed caregiver. Genetic analysis revealed that he had the same mutation as the proband, but he had never been treated with levothyroxine. His basal fT4 level was 0.75 ng/dL, which was below the reference range (0.9–1.7 ng/dL) without an elevation of the TSH level. The basal serum levels of other pituitary hormones were within the reference range. His serum triglyceride (TG) and low-density lipoprotein (LDL) were also within the reference range (Table 1). The TSH response to the TRH stimulation test was insufficient, with a diminished increase in thyroid hormone levels. In contrast to the proband, the TSH response was not prolonged (Fig. 2). Based on these findings, the diagnosis of subclinical hypothyroidism was made, and levothyroxine supplementation was initiated. Once daily 50 μg levothyroxine supplementation was sufficient to maintain his thyroid hormone level within the reference range, but the TSH level was below the reference range (TSH = 0.032 mIU/L, free triiodothyronine [fT3] = 3.43 pg/mL, and fT4 = 1.14 ng/dL).
Female carriers (mother and sister of the proband: II-2 and III-1)
At the time of the study, the mother was 38 years old. Her height was 164.7 cm, weight was 64.6 kg, and BMI was 23.8 kg/m2. She did not have a medical history of thyroid disease, and was previously healthy. She experienced menarche at 15 years of age. She was able to breastfeed her two children, including the proband. Thyroid function tests revealed central subclinical hypothyroidism (TSH = 1.35 mIU/L, fT3 = 2.6 pg/mL [reference range 2.3–4.3 pg/mL], and fT4 = 0.84 ng/dL). The other laboratory results are summarized in Table 1. The mother did not have PRL deficiency. Her serum TG and LDL were within the reference range. Levothyroxine supplementation (25 μg once daily) was sufficient to maintain her thyroid hormone level within the reference range (TSH = 1.00 mIU/L, fT3 = 2.68 pg/mL, and fT4 = 1.10 ng/dL).
The older sister was four years old at the time of the study. Her height was 95 cm (−1.82 SD), and her weight was 15.8 kg (−0.14 SD). Her development was normal, and she had no obvious health problems. Her basal pituitary functions were normal (Table 1). Her TSH response to the TRH test was impaired. The levels of other hormones, including prolactin, were within the reference range for her age (Fig. 2). She was able to maintain normal thyroid hormonal levels without supplementation.
Discussion
The IGSF1 protein contains 12 immunoglobulin domains in two clusters of five and seven motifs, which are separated by a linker segment and followed by a transmembrane and a cytoplasmic region (14). Although more than 15 pathogenic mutations in the IGSF1 gene have been described to date, most mutations exist in the extracellular portion; one frameshift mutation has been reported in the N-terminal domain (15,16). The novel mutation (c.2713C>T, p.Q905X) of the IGSF1 gene found in the present family also resides within the third loop in the extracellular portion. Nakamura et al. reported that the p.R1189X mutation located more distally in the extracellular domain prevents totally IGSF1 protein trafficking to the membrane cell surface, whereas the p.Q645X mutation located in the proximal extracellular domain may cause accelerated RNA decay or unstable peptide degradation (17). The mutation identified here, p.Q905X, is expected to result in a loss of function through one of these mechanisms, resulting in CCH in the proband.
In the proband, the TRH stimulation test showed a diminished increase and delayed decrease in the serum TSH level. In contrast, in the uncle carrying the same mutation, the TSH response to the TRH test was inappropriately low. The TSH responses in previously reported cases were similar to those observed in the uncle (9,16,18). However, one patient with IGSF1 deficiency showed high basal and peak serum TSH levels, in addition to a delayed decrease (17). Although the mutant is likely pathogenic, the uncle carrying the mutation did only develop subclinical hypothyroidism. A recent study in the rat showed that Igsf1 is expressed in the embryonic and adult hypothalamus and in the pituitary gland (GH, TSH, and PRL-producing cells) (12). Thus, CCH patients with inactivating IGSF1 mutations may possess both pituitary and hypothalamic defects because IGSF1 is expressed in both regions, but this needs further characterization.
The proband's mother is a heterozygous carrier of the mutation and was found to have subclinical hypothyroidism. Her laboratory data were negative for thyroid autoantibodies, including antithyroid peroxidase antibodies, antithyroglobulin antibodies, and anti-TSH receptor antibodies. It has been reported that CH is present in one-third of female carriers, many of whom are overweight (13). Subclinical hypothyroidism was also found in the mother of the proband in a previous report (13).
None of the family members, including the proband, presented an obvious prolactin deficiency. According to previous reports, about 70% of patients had a PRL deficiency, and the remaining 30% patients had normal PRL secretion. The reason for this variability remains unclear. The phenotypic spectrum may be associated with other sequence variants (19).
Another important aspect of IGSF1 deficiency is the neurological development. It was previously reported that 24 male patients with hemizygous IGSF1 mutations had no obvious mental retardation, despite delayed diagnosis (13). However, Joustra et al. (20) recently reported that adult male patients exhibited mild deficits in attention control with preserved memory function, and these results were not related to treatment with levothyroxine or the age at which replacement therapy was initiated. In the family reported here, the maternal uncle was not on levothyroxine until adulthood. Although he had a delayed growth spurt, he did not need special education. Detailed neurodevelopmental testing was not performed for this patient, but careful follow-up for neurocognitive functions is necessary to elucidate functional aspects of IGSF1 on the human brain.
IGSF1 mutations can be associated with CCH, postpubertal macroorchidism, hypoprolactinemia, and transient partial GHD (13). The spectrum of clinical manifestations does, however, vary within the family members who are carrying the same mutation, as illustrated also in the present family. Phenotypic variability for the same mutation and variable penetrance and/or expressivity are relatively common in other molecular defects, resulting in combined pituitary hormone deficiency (21). IGSF1 is speculated to affect TRHR signaling and to be involved in postnatal maintenance of the normal function of pituitary thyrotrophs (22). The reasons for the phenotypic variability remain currently unknown but could include an interplay with other sequence variants (e.g., in modifier genes), epigenetic mechanisms, or environmental factors.
In conclusion, a novel mutation (c.2713C>T, p.Q905X) was identified in the IGSF1 gene that is associated with CCH in a Japanese family. Further accumulation of patients with IGSF1 deficiency will be helpful to understand better the clinical spectrum of this disease.
Footnotes
Acknowledgments
We are grateful for helpful comments provided by Dr. Nakamura, Department of Molecular Endocrinology, National Research Institute for Child Health and Development, Japan. This research was partially supported by the Practical Research Project for Rare/Intractable Diseases from Japan Agency for Medical Research and Development, AMED.
Author Disclosure Statement
The authors declare that no competing financial interests exist.