
Abstract
Thyronamines (3-T1AM, T0AM) are endogenous compounds probably derived from L-thyroxine or its intermediate metabolites. Combined activities of intestinal deiodinases and ornithine decarboxylase generate 3-T1AM in vitro. Alternatively, 3-T1AM might be formed by the thyroid gland and secreted into the blood. 3-T1AM and T0AM concentrations have been determined by liquid chromatography–tandem mass spectrometry analysis (LC-MS/MS) in tissues, serum, and cell lines. However, large variations of 3-T1AM concentrations in human serum were reported by LC-MS/MS compared with a monoclonal antibody-based immunoassay. These differences might be caused by strong binding of the highly hydrophobic 3-T1AM to apolipoprotein B100. Pharmacological administration of 3-T1AM results in dose-dependent reversible effects on body temperature, cardiac function, energy metabolism, and neurological functions. The physiological relevance of these actions is unclear, but may occur at tissue concentrations close to the estimated endogenous concentrations of 3-T1AM or its metabolites T0AM or thyroacetic acid (TA1). A number of putative receptors, binding sites, and cellular target molecules mediating actions of the multi-target ligand 3-T1AM have been proposed. Among those are members of the trace amine associated receptor family, the adrenergic receptor ADRα2a, and the thermosensitive transient receptor potential melastatin 8 channel. Preclinical studies employing various animal experimental models are in progress, and more stable receptor-selective agonistic and antagonistic analogues of 3-T1AM are now available for testing. The potent endogenous thyroid hormone-derived biogenic amine 3-T1AM exerts marked cryogenic, metabolic, cardiac and central actions and represents a valuable lead compound linking endocrine, metabolic, and neuroscience research to advance development of new drugs.
Introduction
I
Detection of Endogenous Thyronamines, Biosynthesis, Metabolism, and Distribution
Thyronamine assays in tissues and in blood
Thyronamines (TAM) were synthetized soon after the detection of L-thyroxine (T4) and subsequently tested for their biological or pharmacological action as thyroid hormone (TH) analogues or biogenic amines by several groups studying TH metabolism and action (36 –41). The physiological TAM concentration and the pathways and regulation of TAM biosynthesis have been controversial in the past. While TH were classically determined via chromatographical or immunological methods, liquid chromatography–tandem mass spectrometry (LC-MS/MS) has been initially used for the unequivocal detection of endogenous TAM. 3-T1AM and iodine-free TAM (T0AM) have been the first and, until now, only two of nine potential TAM derivatives identified as endogenous metabolites in mouse tissue and serum using LC-MS/MS (1). The existence of 3-T1AM in human and animal blood has been challenged by the publication of Ackermans et al. (42). Their online solid-phase extraction (SPE) method combined with LC-MS/MS, which had a detection limit of 0.25 pmol/mL, did not detect any endogenous 3-T1AM or T0AM in plasma from rats, or in human plasma or thyroid tissue. They only found 3-T1AM and T0AM in plasma and liver from rats treated with synthetic 3-T1AM and T0AM (42). This publication raised doubts about the endogenous existence and biosynthesis of 3-T1AM. Meanwhile, endogenous 3-T1AM has been detected in various species such as rat, mouse, Djungarian hamster, and humans in serum and tissue samples (Table 1). The differences in the results of LC-MS/MS assays might be related to the strong binding of 3-T1AM to proteins, particularly to apolipoprotein B100 (apoB100) in human serum, which leads to very low free 3-T1AM serum concentrations (43). The procedure of extracting 3-T1AM from samples is analytically highly complex and time-consuming. Therefore, even moderate differences in pre-analytical sample workup and process efficiency in extraction, as well as in the sensitivity of the mass spectrometer, may significantly limit the final yield.
Determined by mass spectrometry.
Measured by immunoassay.
3-T1AM, 3-iodothyronamine.
The relationship between human serum 3-T1AM concentration, TH concentration, and some common clinical chemistry variables have been investigated in a recent study by Galli et al. (4). They measured an average 3-T1AM serum concentration of 0.219 ± 0.012 pmol/mL in euthyroid individuals (n = 22). The 3-T1AM concentration was significantly correlated to serum concentrations of total T4, total triiodothyronine, glycated hemoglobin, brain natriuretic peptide, and γ-glutamyl transpeptidase (4). In a small cohort of patients, this group showed that 3-T1AM was significantly higher in diabetic (n = 7) compared with non-diabetic patients (n = 18) and significantly correlated with HbA1c concentrations, suggesting a potential role of 3-T1AM in insulin resistance (4). Despite the clear cardiac effect of 3-T1AM in various experimental models (1,22,35,44 –46), the authors could not find any significant difference in patients with cardiac dysfunction (4).
A recent study reported on concentrations of 11 TH and TAM in amphibian sera (Rana catesbeiana) using tandem mass spectrometry (47). Their methodological approach extended previous work (48) and detected 3-T1AM concentrations (3.11 ± 1.18 nmol/L) higher than those of T3 (1.15 ± 0.22 nmol/L) and similar to T4 (7.68 ± 2.45 nmol/L) in a concentration range for 3-T1AM similar to that reported for human serum (5,6). However, inspection of details of this method as published indicates some shortcomings among the extensive and careful elaboration of quality control parameters of both the pre-analytical and the analytical parts of the method. In contrast to common established operating procedures and practice in quantitative mass spectrometry, the authors did not report an inclusion of a stable isotope-labeled standard for 3-T1AM, but rely on those for T4, T3, and rT3 for TAM quantification. This analytical aspect is of relevance considering the experience of the authors and other groups (Scanlan et al. and Saba et al., pers. commun.) that the LC-chromatographic 3-T1AM peak frequently is superimposed by an abundant mass interfering with 3-T1AM quantification and recovery due to matrix effects impairing process efficiency with potential impact on its quantification.
Up to now, the concentrations of 3-T1AM reported using LC-MS/MS in serum ranged from 10–12 M (pM) to 10−9 M (nM) due to different pre-analytical extraction methods and LC-MS/MS device-dependent limits of detection. To avoid these analytical problems, a highly specific monoclonal antibody-based 3-T1AM chemiluminescence immunoassay (CLIA) was developed to provide an independent analytical tool for 3-T1AM measurement in human serum (5,6). As the concentration range of 3-T1AM using the LC-MS/MS is much lower (by about two orders of magnitude) compared with the data obtained with the CLIA (14–66 nM in healthy individuals, cardiac patients, and patients with non-thyroidal illness) (5,6), doubts have arisen about the specificity of this antibody. Many TH-related compounds have been tested for their potential cross-reactivity, but none of those derivatives with notable cross-reaction have been detected so far in human serum at relevant concentrations. LC-MS/MS analysis of immune-extracts of human serum prepared by this monoclonal antibody revealed only 3-T1AM as bound ligand (C.S.H. and J.K., unpublished data). Lately, several new TAM derivatives have been detected (27), which have not yet been systematically tested as ligands in this CLIA. Since the CLIA assay is based on the competition between endogenous 3-T1AM and a labeled 3-T1AM reporter, another crucial issue is the availability of a calibration curve obtained in the presence of the same binding proteins that are present in serum, particularly apoB100 (43). It might well be that endogenous 3-T1AM does not fully equilibrate with the exogenously added 3-T1AM tracer in this assay setup. On the contrary, pre-analytical steps required for sample preparation for the LC-MS/MS analytics might not completely extract all endogenous 3-T1AM from its serum binding sites. Furthermore, internal standards added to the samples of interest might not fully equilibrate with endogenous protein-bound 3-T1AM and thus mislead readouts of the isotope dilution method on which LC-MS/MS quantification is based. Binding equilibria and extraction efficiency might be affected by matrix composition of the various assay formats used in pre-analytics for LC-MS/MS (i.e., extraction with organic solvents and protein precipitation of diluted serum samples). In contrast, the CLIA is performed with serum samples diluted in bovine serum albumin containing phosphate-buffered saline or human serum in which the classical TH T4 and T3 have been depleted by dextran-coated charcoal or ion exchanger treatment. Both procedures will alter composition of sera compared to the native serum sample. Up to now, no natural or commercial serum matrix is available devoid of endogenous 3-T1AM for analytical comparison of various analytical procedures so far employed in 3-T1AM analytics.
In aggregate, although the existence of 3-T1AM in serum and tissue has been shown several times since 2004 (Table 1), controversies still exist about the appropriate method of detection as well as serum and tissue concentrations of endogenously present and exogenously administered 3-T1AM in body fluids and tissues. Reproducible protocols using a pre-analytical extraction and LC-MS/MS may only provide the free or at least a fraction of non-apoB100 bound 3-T1AM, while the CLIA without any pre-analytical extraction is a promising technique to determine total 3-T1AM. However, the LC-MS/MS approach has been a useful tool to study the endogenous presence, the metabolism, and biosynthesis of 3-T1AM in cell culture experiments (3,49 –52). Recently, an intensive LC-MS/MS validation of a method to measure nine TH and six TAM in parallel from a single cell culture media sample has been published (53).
TAM biosynthesis
T4AM, 3,5-diiodothyronamine (3,5-T2AM), T0AM, and several N-or O-acetylated T4 derivatives (54) have been synthesized soon after the discovery of “thyroiodine” (55), the isolation (56), identification, and chemical synthesis of T4 (57 –59). These compounds have been tested in various in vivo animal bioassays (tadpoles, axolotls, rats) with rather divergent results concerning growth, metamorphosis, oxygen consumption, and other endpoints of action (36,37,54,60).
TAM differ from TH only by the absence of the amino acid carboxyl group at the alanine side chain. Similar to TH, there are nine possible TAM derivatives regarding the number or the position of iodine atoms. However, up to now, only two TAMs—the iodine-free T0AM and the mono-iodinated 3-T1AM—have been detected in vivo. The presence of any other endogenous TAM in vivo has not yet been reported. Although the biosynthesis of 3-T1AM from TH appears likely, the sequence of reactions, organs, as well as cellular locations of synthesis are not finally defined.
De novo biosynthesis of 3-T1AM would require partial oxidative iodination and ether-bond coupling of two tyrosyl rings resembling the biosynthesis of TH, which solely occurs bound to their precursor protein thyroglobulin (61). Alternatively, de novo biosynthesis of 3-T1AM from T0AM would require iodination of T0AM. So far, both the iodination and the coupling reactions of tyrosyl residues have been described only within the thyroid gland where they occur in the specialized luminal compartments of the thyroid angiofollicular unit (62) and involve thyroperoxidase (TPO) and dual oxidase (63). However, neither significant release of TH with lower iodination grade than T4 or T3 nor direct secretion of 3-T1AM from the thyroid gland has been reported, supporting the hypothesis of an extrathyroidal TAM biosynthesis from TH (5).
Assuming that 3-T1AM is a downstream metabolite of T4, the biosynthesis would require the decarboxylation of the phenylalanine side chain in combination with deiodination by removing up to three iodine atoms. A systematic in vitro screen of all possible TAM deiodination reactions revealed that all three deiodinases—Dio 1, 2, and 3—catalyze TAM metabolism with each isozyme exhibiting a unique substrate specificity. These data contribute to confining the biosynthetic pathways for potential 3-T1AM and T0AM biosynthesis (52). Since 1974, it has been proposed that the ubiquitous enzyme aromatic L-amino acid decarboxylase (AADC, E.C. 4.1.1.28) mediates T4AM biosynthesis via decarboxylation of T4 (40). This hypothesis is supported by the broad substrate specificity of AADC and the structural similarity of TAM to biogenic amines, also called monoamines (64). AADC, also known as L-DOPA decarboxylase (DDC), requires pyridoxal-5-phosphate as a co-factor during the conversion of L-DOPA to dopamine or of 5-hydroxytryptophan (5-HTP) to serotonin (65). In addition, AADC is also considered to be involved in the biosynthesis of trace amines such as tryptamine, and β-phenylethylamine (β-PEA) (66,67). In 2012, this hypothesis was tested and refuted. It has been shown by in vitro studies that AADC does not accept TH as substrates to generate 3-T1AM (7). Moreover, four patients with AADC deficiency (OMIM #608643), a rare congenital disease with fewer than 100 cases reported worldwide, exhibit normal 3-T1AM serum concentrations (7). Thus, the responsible decarboxylase was still elusive. The first description of TAM biosynthesis from TH comes from cell culture experiments. It has been shown that 3-T1AM is generated in vitro in H9c2 rat cardiomyoblasts exposed to T3 (3), a finding that has not been confirmed so far by independent studies. In contrast, no 3-T1AM production was observed in cultured primary rat thyrocytes and rat FTRL-5 thyrocytes offering T4 as substrate (49). Therefore, it seems reasonable to assume that 3-T1AM biosynthesis occurs in peripheral tissues independent of the thyroid gland as the TH producing organ.
Indeed, a study in T4-substituted thyroid cancer patients after thyroidectomy/radioiodine treatment lacking functional thyroid tissue revealed 3-T1AM concentrations similar to or even slightly higher than in healthy thyroid-intact individuals as assessed by the 3-T1AM immunoassay, suggesting mainly extrathyroidal 3-T1AM production from administered T4 or its deiodinated metabolites (5). However, a recent study using isotope-labeled T4 in hypothyroid mice to study 3-T1AM biosynthesis from TH speculated that 3-T1AM might not be an extrathyroidal T4 metabolite, as its biosynthesis seems to be dependent on the sodium–iodide symporter (NIS) and TPO (8). This assumption is based on the observation that in these hypothyroid mice treated with labeled T4, the LC-MS/MS analysis of liver extracts detected labeled T3 but not 3-T1AM, suggesting T4 to T3 conversion but no further metabolism to 3-T1AM. To make these mice hypothyroid, their drinking water was supplemented with 0.1% methimazole and 0.2% potassium perchlorate to inhibit both the thyroid enzyme TPO and the NIS, respectively. This classical model of pharmacologically induced hypothyroidism would produce a systemic inhibition of TPO and NIS, but might also affect deiodinating and decarboxylating enzymes involved in 3-T1AM biosynthesis (9), whereas surgery and/or radiotherapy as used for thyroid cancer treatment would exclusively affect the thyroid gland itself. Based on these observations, it was hypothesized in a following study that intestinal passage might be an important step in 3-T1AM biosynthesis, since thyroid cancer patients received T4 orally (5) whereas the hypothyroid mice in the study using labeled T4 were injected i.p. with T4 (8). Using the mouse everted gut sac model in combination with LC-MS/MS, it was demonstrated that 3-T1AM production from T4 is possible in mouse intestine via several deiodination and decarboxylation steps (9). Gene expression analysis confirmed the expression of all three deiodinases, as well as the high transcript concentrations of ornithine decarboxylase (ODC, E.C. 4.1.1.17) in mouse intestine. Subsequent experiments utilizing purified human ODC revealed that this enzyme, in contrast to human AADC (7), can mediate decarboxylation of T4 and 3,5-T2 to the respective TAM, demonstrating that the intestine expresses the entire molecular machinery required for 3-T1AM biosynthesis from T4 (9) (Fig. 1). Interestingly, it has been shown that mRNA expression of enzymes involved in TAM production, that is, Odc and Dio, is strongly decreased by pharmacological treatment with methimazole and potassium perchlorate independently of thyroid status, limiting the validity of the respective hypothyroid mouse models in the context of unravelling biosynthesis of TAM (9).
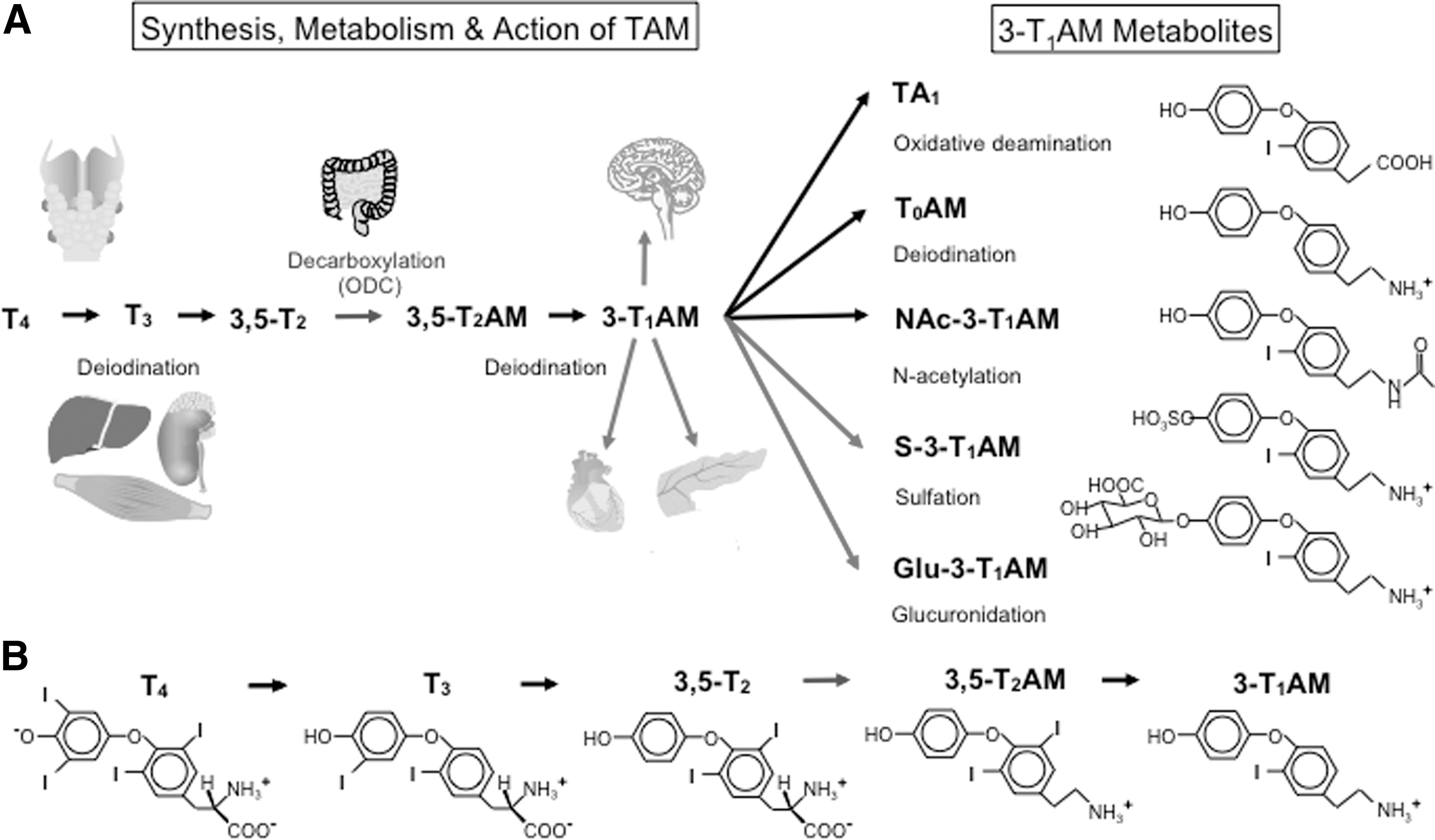
Synthesis, metabolism, and action of 3-iodothyronamine (3-T1AM). (
Hence, intestinal 3-T1AM biosynthesis from T4 involving decarboxylation by Odc with subsequent deiodination can explain the apparent discrepancy between 3-T1AM serum concentrations in thyroid cancer patients substituted orally and hypothyroid mice injected i.p. with T4. Whether this Odc-mediated 3-T1AM biosynthesis pathway is also present in other tissues and if further amino acid decarboxylases are also involved in this process need to be further tested in the future.
Endogenous metabolites
It has been demonstrated in vitro, ex vivo, and in vivo that 3-T1AM can rapidly be metabolized via deiodination to T0AM (49,50,52), oxidative deamination followed by aldehyde oxidation to TA1 (2,49 –51), N-acetylation to (N-Acetyl-3-iodothyronamine) NAc-3-T1AM (8), or in conjugation with sulfate to 3-T1AM-sulfate (68) or glucuronide to 3-T1AM-glucuronide (8) (Fig. 1). The existence of endogenous T0AM has already been shown in 2004 by Scanlan et al. (1). Besides T0AM, TA1 is the major metabolite of 3-T1AM metabolism and the deiodinated degradation product of triiodothyroacetic acid (Triac, TA3) (27,51,69). Both, T0AM and TA1, have been found in trace amounts in human serum and mouse brains (2,29), and might exert some of the effects so far ascribed to 3-T1AM after 3-T1AM administration in mice.
Using LC-MS/MS, Hackenmueller et al. showed that a single injection of 3-T1AM (25 mg/kg, i.p.) in mice can lead to the formation of all abovementioned 3-T1AM metabolites within minutes to hours, as analyzed in brain, kidney, liver, WAT, and serum (8). In this study, one of the interesting newly found metabolites after 3-T1AM injection was the lipophilic compound NAc-3-T1AM, since peripheral N-acetylation has not yet been described for any TH derivative. Acetylation is a common modification for biogenic amines, polyamines, amino sugars, and proteins (70 –72). Thus, NAc-3-T1AM could constitute a relevant downstream metabolite mediating some of the 3-T1AM effects. Furthermore, NAc-3-T1AM might be detected by the 3-T1AM CLIA, but may not interfere with LC-MS/MS-based determination of serum 3-T1AM, as long as NAc-3-T1AM does not decompose spontaneously during the steps of pre-analytic extraction and LC-MS/MS detection.
Serum distribution, cellular uptake, and storage
3-T1AM does not bind the same serum proteins as TH. Whereas TH strongly bind to TBG, TTR, and albumin, but only to a minor extent to lipoproteins (73,74), a high fraction of 3-T1AM (> 90%) is specifically non-covalently bound to apoB100 (43). The dissociation constant (Kd) of 17 nM results in a low free 3-T1AM concentration in serum (43). The apolipoprotein occurs in plasma in two main isoforms: apoB100 and apoB48. In humans, apoB100 is expressed in the liver and is present on very low density lipoprotein and low density lipoprotein (LDL), whereas apoB48 is expressed in the intestine and is present on chylomicrons and their remnants (75).
The human circulating apoB100 concentration is 1.5–3.0 μM (7–153 mg/dL), depending on the lipid status, which is regulated by nutrition, hormonal influences, metabolic disorders, and genetic background (76). ApoB100 serves as a ligand for the LDL receptor (LDLR) (Fig. 2), which is mainly responsible for the removal of LDL from plasma. The receptor-mediated endocytosis involving apoB100 is a well-established cellular uptake mechanism for small molecule lipids such as cholesterol and triglycerides (76). Hence, the physiological role of the strong binding of 3-T1AM to apoB100 may be to provide a mechanism for transportation and entry of 3-T1AM into target cells via LDLR mediated endocytosis (43).
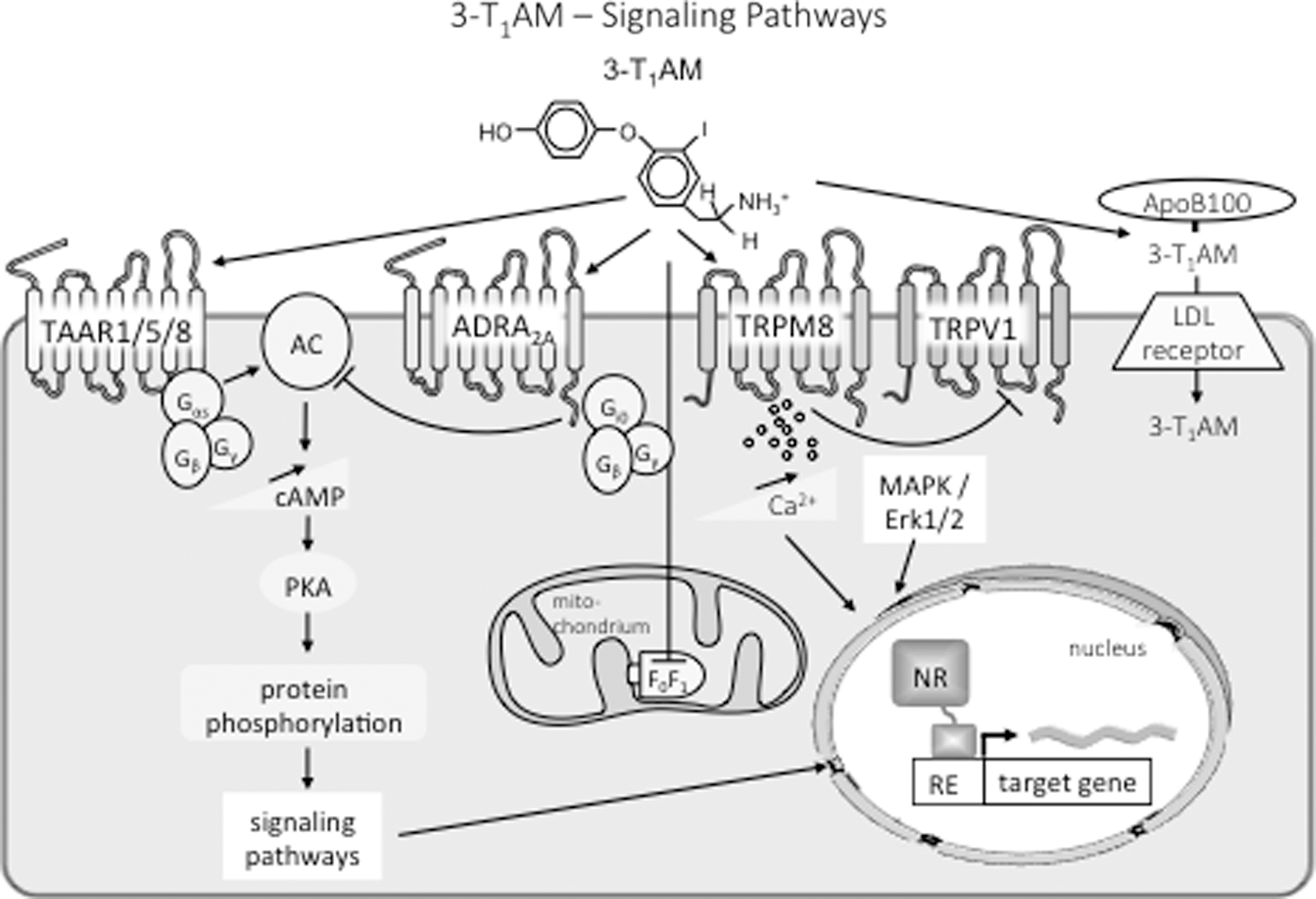
Summary of known cellular targets and signaling pathways for 3-T1AM. The TH-derived biogenic amine 3-T1AM acts via TAAR to stimulate adenylyl cyclase and protein kinase A signaling pathways or via inverse agonistic action on the α-adrenergic receptor 2A and Gi0 cascade. Stimulation of the transient receptor potential melastatin 8 channel increases intracellular calcium and blocks action of the transient receptor potential vanilloid 1 channel. Modulation of MAPK/Erk1/2 pathways has also been reported. 3-T1AM also interferes with mitochondrial function via modulation of the F0F1 ATPase activity. In blood, 3-T1AM binds with high affinity to the apolipoprotein B100 and may enter cells via low-density lipoprotein receptor-mediated endocytosis. Further cell membrane transporters for the biogenic amine 3-T1AM may exist.
Systemic administration of 3-T1AM permits its passage through the blood–brain barrier, but it is unknown by which precise mechanism this occurs. Remarkably, 3-T1AM is not a substrate of TH transporters such as MCT8 or MCT10 (1,77 –79). To identify putative 3-T1AM transporter(s), a systematic large-scale screening analysis of the solute carrier transporter family was performed. No single specific TAM transporter was identified from this screen. However, sodium- and chloride-independent, pH-dependent, TAM-specific cellular uptake may involve multiple transporters (77,78). An apparent sodium-dependent 3-T1AM uptake has been shown in cardiac H9c2 cells (3).
The uptake of exogenous radiolabeled 125I-3-T1AM from the bloodstream revealed that 3-T1AM is systemically distributed to various tested organs (80). A significant time-dependent increase in tissue versus blood concentration occurred in the gallbladder, stomach, intestine, liver, and kidney, reflecting a biliary excretion and enterohepatic recycling of 3-T1AM at those concentrations administered (80,81). These radiotracer studies revealed that liver, muscle, and adipose tissues might be regarded as 3-T1AM storage sites. However, no distinction between 3-T1AM and its rapidly formed metabolites can be made from these radioactive measurements.
Inactivation and termination of TAM action by monoamine oxidases and conjugating enzymes
Oxidative deamination of TH generates iodoacetic acids (TA), tetraiodothyroacetic acid (Tetrac; TA4), Triac, and diiodothyroacetic acid (Diac, TA2), and so on, which are habitually present at low concentrations in human serum (69,82 –85). Even though oxidative deamination has been described as inactivating pathway for monoamines, Triac has significant thyromimetic activity. In vitro, it is more potent than T3 for transcriptional regulation by TRβ1 and TRβ2 isoforms, while regulation by TRα1 is equivalent for both ligands (86,87). Interestingly, the cephalochordate Branchiostoma floridae, a marine invertebrate living in warmer coastal areas, expresses a TH receptor, which is activated by Triac but not by T3 (88). Moreover, a naturally occurring non-selenodeiodinase catalyzing 5-deiodination of Tetrac and Triac has been identified in amphioxus. These findings support the hypothesis that Triac is a primordial bioactive TH (89). It has been shown that TAM are substrates of monoamine oxidases (MAO) and semicarbazide-sensitive amine oxidase (SSAO), leading to the formation of the representative TA (3,51). Hence, MAO inhibitors (e.g., iproniazid, pargyline, and semicarbazide) have been used in various in vitro experiments to distinguish TAM from TA action (2,29,51).
Sulfation and glucuronidation are so-called phase II detoxication reactions aimed to increase the water solubility of the TH and arylamines to facilitate their biliary and/or urinary clearance (90). The concentrations of TH sulfate in plasma, bile, and urine are normally very low because of the rapid degradation of these conjugates by Dio1. In contrast to the TH sulfates, TH glucuronides are efficiently excreted in the bile. However, after intestinal hydrolysis of the TH glucuronides by bacterial β-glucuronidases, part of the liberated TH is reabsorbed and again biologically active, constituting an enterohepatic cycle (90,91). Pietsch et al. have reported that sulfation contributes to the metabolism of TAM in human liver and that sulfotransferase (SULT) activities may regulate the physiological effects of hypothermia and decreased cardiac output shown in mice (1) after administration of exogenous 3-T1AM (68). Using a radiochemical enzymatic assay for human liver SULT activities, they identified SULT1A3 as the major contributor to sulfation of T0AM and 3-T1AM, whereas SULT1A1 was the major contributor to sulfation of T3AM (68). Ether bond cleavage, which has been reported to contribute to TH degradation during sepsis and non-thyroidal illness and oxidatively liberating iodine (69), has not been detected so far for 3-T1AM.
Receptors and Signal Transduction
TAAR1 and other TAARs
TAARs comprise a family of GPCR activated by endogenous aminergic metabolites of aromatic amino acids and several psychostimulants, and are involved in odor perception (92,93). These promiscuous receptors react to a broad spectrum of mainly volatile ligands, show significant species differences, and share homology with biogenic amine GPCR. Only few selective and isoform-discriminative ligands have so far been identified (94). TAAR1, a receptor for amphetamine and methamphetamine, has initially been proposed as the main target of 3-T1AM (1), but meanwhile other TAAR such as TAAR5 and TAAR8 are considered as potentially relevant targets based on in vitro studies using cellular receptor overexpression models (Fig. 2) and the in vivo observation that the hypothermic and several metabolic effects of pharmacological 3-T1AM administration persisted in Taar1 knockout mice (95). Furthermore, as indicated below, in vitro effects of 3-T1AM stimulating TAAR1-dependent Gs-mediated cAMP production contrast with most of the observed effects in cardiac models for 3-T1AM action, which are mainly inhibitory. Therefore, other family members were examined as potential 3-T1AM targets either based on their tissue specific expression patterns (e.g., TAAR8 isoforms in the heart) or their so far known ligand binding and structural properties (TAAR5). Orthologues to the mammalian Taar1 with high sequence variations were identified in many species. However, vertebrate as well as mammalian orthologues analyzed exhibited high conservation for 3-T1AM as potent ligand with EC50 values in the range of 0.75–2.4 μM, while rat Taar1 excelled with a low EC50 of 0.1 μM (96). Ligand properties of two other trace amines tested, β-PEA and p-tyramine, were also highly conserved but did not show this distinct property of a lower EC50 in rat versus mouse Taar1. These authors interpreted their observations as evidence for distinct species differences in expression of TAAR repertoires, but high conservation of the ligand binding pocket for promiscuous biogenic amines, including 3-T1AM, which by itself is a multi-target ligand (96). Surprisingly, Taar1 has been localized to the primary cilium structure of the apical pole of rat thyrocytes using immunohistochemical and cell-biological tools (97). The impact of this observation with respect to a potential sensor function for 3-T1AM or other biogenic amines via Taar1 at the cilia at the inner surface of the angiofollicular unit of thyroid follicles remains to be clarified, especially in its potential context for TH biosynthesis.
Taar5 interactions with 3-T1AM and GPCR were also studied in more detail. In mouse brain, Taar5 and Taar1 exhibit in part overlapping expression patterns in regions relevant for metabolic and behavioral effects ascribed to 3-T1AM action, such as the ventromedial hypothalamus, amygdala, and others, and Taar5 is the most conserved subtype among mammalian species (14). Comparative analysis of signaling properties of human and mouse Taar5 using HEK cell in vitro expression models revealed evidence that human TAAR5 exhibits inverse agonistic properties after 3-T1AM stimulation in contrast to mouse Taar5, which did not show such a response. This might have therapeutic relevance, but such comparisons have not been made yet using neuronal cell models. If these properties also apply to in vivo conditions, the Taar isoform mediating the hypothermic and metabolic effects of 3-T1AM in mice remains elusive. Both human and mouse Taar5 show marked basal activity for the Gq/11 signaling pathway, while mTaar5 but not hTaar5 display basal Gs activity in transfection models. Such behavior might be relevant for functional hetero-oligomer formation with adrenergic GPCR or other membrane receptors interacting with TAAR and 3-T1AM as one of its endogenous ligands.
Adrenergic receptors and other GPCRs
In a first attempt to characterize the mode of action of TH-derived TAM, binding and competition studies were performed with the synthetic ligand 3,3′,5-triiodo-L-thyronine (Triam) using turkey erythrocyte membranes as a source rich in β-adrenergic receptors (ADR) (38,98). Triam, in contrast to tyramine and the classical TH T4, T3, and rT3 proved as a ligand equipotent to isoproterenol in competition for (-)(3H)-dihydroalprenolol binding and blocked cAMP formation in functional assays. Similar effects were obtained using 3,5-diiodotyramine, 3,5-diiodothyronamine, and T0AM; 3-T1AM was not tested (38). No receptor studies were performed using T4AM, the first TAM to be identified 90 years ago (36,37).
Recently, the interaction of the endogenous ligand 3-T1AM with β1- (ADRβ1) and β2- (ADRβ2) adrenergic receptors was studied in detail (99) employing transfected HEK293 cells and human conjunctival epithelial cells (IOBA-NHC), which express endogenous ADRβ (Fig. 2). While 10 μM 3-T1AM concentrations alone showed only weak activation in comparison with 1 μM isoproterenol on both ADRβ using a label-free dynamic mass redistribution assay as readout, 3-T1AM at 1 μM concentration enhanced the isoproterenol binding and Gs-mediated cAMP stimulation at ADRβ2 but not ADRβ1 signaling and its cAMP response to 0.1 mM norepinephrine. In IOBA-NHC cells, timolol, the non-selective blocker of ADRβ, inhibited Ca2+ influx stimulated by both 1 μM norepinephrine and by 10 μM 3-T1AM, indicating effective interaction with endogenous ADR in a non-transfected human cell line.
Persistence of metabolic and thermoregulatory effects of 3-T1AM in TAAR1 knockout mice (95) indicated the contribution of other receptors including GPCR in 3-T1AM signal transduction. Employing the dynamic mass redistribution assay technology and cells transfected with either ADRα2a or TAAR1 alone or in combination revealed clear Gi/o-mediated activation at ADRα2a by 10 μM 3-T1AM and concentration-dependent inhibition of norepinephrine-induced MAPK (ERK1/2) activation (10). In HEK293 cells expressing ADRα2a/TAAR1 hetero-oligomers, 3-T1AM uncoupled Gi/o signaling without interfering with MAPK activation by norepinephrine. Thus, 3-T1AM might act as a partial agonist at ADRα2a but antagonistic with respect to AMPK activation by norepinephrine. However, these interesting in vitro observations contrast with different dose-dependent outcomes of in vivo experiments with respect to regulation of glucose homeostasis by β-cells in mouse Langerhans islets, which endogenously co-express ADRα2a and Taar1 at high concentrations. Regard et al. (100) proposed 3-T1AM-induced Gi/o-mediated signaling via ADRα2a in pancreatic β-cells in their experiments on regulation of glucose homeostasis and thermoregulatory effects exerted by 3-T1AM in transgenic mice, where Gi/o-signaling was compromised by expression of pertussis toxin. They injected a single 50 mg/kg dose of 3-T1AM, resulting in inhibition of insulin secretion, and they interpreted this finding as ADRα2a-mediated Gi/o activation (100). However, repeated administration of 5 mg/kg of 3-T1AM in male mice for six days did not alter parameters of glucose homeostasis (fasting basal blood glucose and glucose tolerance test), hepatic glycogen, food intake, body temperature, or expression of selected hepatic genes involved in glucose homeostasis (10).
These controversial outcomes of two in vivo experiments differing in dose and duration of 3-T1AM administration suggest either additional factors are involved in hormonal regulation of glucose homeostasis or a distinct central versus peripheral dose-dependent mode of action of 3-T1AM.
As previously observed for TAAR isoforms, the ligand binding site of ADRβ is also quite promiscuous and binds ligands such as 3-T1AM, related TAM, and biogenic amines (101). The classical trace amines tyramine and β-PEA act as partial allosteric antagonists for isoproterenol in co-stimulation experiments using cells transfected with ADRβ1/2. This is different for octopamine, which functions as a partial orthosteric ADRβ2-antagonist and ADRβ1-agonist in isoproterenol co-stimulation experiments (101). These observations indicate a delicate balance between catecholamine and trace amine ligand exposure of target cells and cell type-specific co-expression of ADR and TAAR, which might alter their binding and signaling properties if functional heterodimeric receptors are formed. The previously mentioned antagonistic effects of TAM on ADRβ in turkey erythrocyte membranes (98) might illustrate such effects.
Similarly, the complex picture of effects of 3-T1AM in different models studying cardiac action and targets also implies dose-dependent interactions between TAAR forms and classical adrenergic GPCR involved in regulation of cardiac function (see Neuromodulation section below). At this point, 3-T1AM has been proven as a multi-target ligand for various GPCR subclasses acting in (partial) agonistic or antagonistic mode in the presence of their bona fide ligands. But one needs to be aware that most of these in vitro, ex vivo, and in vivo experiments have been performed using rather high 3-T1AM concentrations, while only a few of those effects also persist at lower endogenous nanomolar 3-T1AM concentrations. In addition, 3-T1AM metabolites might rapidly form in vitro in the absence of the high affinity binding protein apoB100, which efficiently protects and stabilizes 3-T1AM in vivo. However, to date, these metabolites have rarely been tested for their interaction and modulation of ADR function.
TRP receptor channels
TRP channels represent a further interesting target for TAM action (Fig. 2). Members of the two major families of TRP channels are ubiquitous rather non-selective cation channels in the plasma membrane acting as cellular sensors for various physical (e.g., cold, heat, pressure, volume), biochemical, and nutritional (e.g., spices, micronutrients) stimuli. Recently, 3-T1AM was characterized as potent activator of the cold-sensitive TRP melastatin 8 (TRPM8) Ca2+ channel expressed in several cell types and involved in cold and pain sensation (15,16). Concentration-dependent activation by 3-T1AM (0.1–10 μM) occurred at much lower IC50 values than reported for its classical stimulants icilin (500 μM) and menthol (15–60 μM). The TRPM8 antagonist BCTC effectively blocked the 3-T1AM effect as illustrated by patch clamp techniques in transfected cells and human cell lines. Of clinical interest is the strong inhibition of the activation of the heat-sensitive TRP vanilloid 1 (TRPV1) channel, that is, the capsaicin receptor, by 3-T1AM-induced TRPM8 activation. Such an inverse mode of 3-T1AM action via TRPM8 could effectively block TRPV1-induced production of proinflammatory cytokines after stimulation with capsaicin or hyperosmolarity (15,16). Thus, 3-T1AM may represent a lead compound in the development of agents interfering with TRPV1 activation related to inflammatory processes or pain sensation. Again, these actions on TRPM8 still require low micromolar 3-T1AM concentrations, and whether this observation relates to TRPM8-mediated sensing of low environmental or low body temperature, such as induced by pharmacological 3-T1AM application, requires further studies. In theory, such an effect of 3-T1AM via TRPM8 on intracellular Ca2+ flux might be part of a cellular feedback loop if functional also at endogenous nM 3-T1AM concentrations.
Additional binding sites: apoB100, mitochondria, and transporters for biogenic amines
In addition to the receptors described above, 3-T1AM might have additional molecular targets. ApoB100 has a single high-affinity binding site for 3-T1AM (43). Since apoB100 concentration in serum lies in the micromolar range, its binding capacity is high, and might account for the technical difficulties of serum 3-T1AM assay, as discussed earlier.
In general, the existence of a high-affinity binding site is related to an important biological function. However, at present, the functional implications of 3-T1AM binding to apoB100 are not clear. In vitro, 3-T1AM increased LDL secretion by HepG2 cells (a neoplastic liver cell line) by about twofold, and slightly favored (by about 20%) LDL uptake by fibroblasts overexpressing LDL receptors. However, in mice fed a high-cholesterol diet and in transgenic mice overexpressing apoB100, subacute 3-T1AM administration (up to 0.4 mg/kg i.p. daily for 15 days) did not modify serum triglycerides, LDL cholesterol, or high-density lipoprotein cholesterol (43). On the other hand, a recent preliminary report (102) showed that total serum cholesterol was indeed reduced in obese mice treated for a week with higher 3-T1AM doses (10 mg/kg daily), although this effect might be related to changes in the expression of several genes involved in lipoprotein metabolism (103).
It has also been hypothesized that LDL endocytosis may be a mechanism of cellular 3-T1AM uptake. The presence of LDL is not necessary for 3-T1AM uptake, which has been observed in vitro in several cells types cultured in the absence of serum proteins (3,49,50). However, in human liver hepatocellular carcinoma cells (HepG2) overexpressing LDL receptors, 3-T1AM uptake increased by about 50% in the presence of exogenously added LDL (43).
Interesting but so far rather confusing data have been reported with regard to the existence of mitochondrial TAM targets or binding sites (Fig. 2). In rat liver mitochondria, 3-T1AM decreased oxygen consumption and increased the release of hydrogen peroxide (18). These actions were observed at concentrations ≥10 nM and were shared by T0AM, which had a similar or higher potency. Using specific inhibitors of respiratory chain complexes, but no concomitant blockade of action of monoamine oxidases, it was concluded that 3-T1AM exerted its action on Complex III. In another investigation (17), low concentrations of 3-T1AM (50 nM) increased ADP-stimulated respiration in H9c2 cells, and activated the F1-ATPase in submitochondrial particles, most likely by inducing the dissociation of IF1, an endogenous inhibitor protein of mitochondrial ATP synthase. Molecular docking analysis also suggested the presence of a putative 3-T1AM binding site overlapping the IF1 binding site. The response to 3-T1AM was, however, biphasic, since in submitochondrial particles micromolar 3-T1AM inhibited either direct or reverse ATPase activity (i.e., either ATP synthesis or ATP hydrolysis). The latter result was confirmed with purified F1-ATPase, but the IC50 was in the order of 20–30 μM.
On the whole, 3-T1AM appears to be able to modulate mitochondrial function, although no specific target sites have yet been identified by binding, competition, or displacement experiments. Inhibition of respiratory activity and/or ATP synthesis occurs at micromolar concentrations, and might contribute to the hypothermic effects described in mice after treatment with high doses of exogenous 3-T1AM (50 mg/kg) (1,23,104). The in vitro responses reported with nanomolar 3-T1AM concentrations are intriguing, but they are still unclear with respect to their molecular mechanisms, functional implications, and potential physiological relevance.
3-T1AM also binds to human dopamine transporter (DAT), as illustrated in cellular HEK transfection models, and inhibited dopamine and norepinephrine (NET) uptake with IC50 in the low micromolar range using striatal synaptosomal preparations of wildtype and TAAR1 knockout mice (95,105). The authors interpreted these findings as evidence that TAAR1 is not involved in the hypothermic effect induced by administration of high pharmacological concentrations of 3-T1AM and that 3-T1AM is selective with respect of inhibition of dopamine and norepinephrine but not serotonin transport (95). Apart from their interference with the plasma membrane transporters, DAT and NET, 3-T1AM also interact with the vesicular membrane transporter VMAT for biogenic amines such as serotonin with an IC50 of 3.2 μM (105). Here, not only 3-T1AM but several of the TAM carrying none, one, or two iodine atoms, or in case of Triam even three iodines, competed effectively with serotonin in contrast with T4, T3, T4AM, and rT3AM, which were ineffective. No evidence was reported that TAM are also substrates for VMAT, providing these agents a unique position among the other endogenous biogenic amines, which act as competitive substrates for VMAT. Some early (indirect) evidence points to a possibility of intracellular receptors such as TAAR1 or functional target sites for 3-T1AM (94,106). Recently, intracellular “intracrine” GPCR signaling has been documented for several receptors, for example after internalization or during recycling to the plasma membrane using life-imaging approaches, caged ligands, and optogenetics (107). Such 3-T1AM-dependent signaling would require transport proteins in the plasma membrane supplying such intracellular receptors with ligands, again a possibility to be studied in more detail.
Major cellular signaling pathways
The biogenic amine 3-T1AM is a highly hydrophobic but positively charged ligand interacting with cell surface receptors and/or intercellular target sites (summarized in Fig. 2). As already indicated, both adrenergic GPCR as well as members of the TAAR family accept 3-T1AM as ligand, but up to now, most of the pathways modulated by receptor interaction have been studied in vitro using rather artificial models such as overexpression of low abundance receptors in heterologous cells. However, some preliminary conclusions can be drawn with respect to intracellular signaling cascades. TAAR couple to various subtypes of G proteins, and both stimulation and inhibition of cAMP production has been reported (14,96,108 –110) while no productive receptor interaction of 3-T1AM with TAAR8 and TAAR8b and related downstream signaling cascades, supposed to mediate some of the in vivo cardiac effects of 3-T1AM, were found. 3-T1AM interaction with TAAR5 causes Gi/q11 activation, resulting in an inverse agonist function compared with ligand norepinephrine (14).
Apart from TAAR interactions, 3-T1AM modulates ADR function, and the main effects reported so far deal with its activation of ADRα2a via Gi/q11, resulting in inverse agonistic action or interaction with ADRβ. Such effects have been delineated in vitro using recombinant expression systems in transfected cells combined with analysis of cAMP formation, Ca2+ signaling, as well as reporter systems for cAMP formation, Ca2+ signaling, and MAPK/Erk1/2 activation. The available investigations point to remarkable species differences in response, distinct competition patterns with bona fide receptor ligands, and remarkable promiscuity in ligand preference (1,10,13,46,95,99,100,108,109,111).
3-T1AM-dependent stimulation of Ca2+ fluxes as demonstrated by patch clamp or Ca2+-fluorophore imaging in ex vivo rodent cardiac models (45) or in cell culture models (15,16) based on endogenous expression of Taar isoforms or adrenergic GPCR supported the relevance of several of these so far identified signaling pathways, although quite high concentrations of 3-T1AM are required for these endpoints. Observations on 3-T1AM-dependent activation of tyrosine kinase and/or phosphatase signaling pathways in rat cardiac models have also been reported (44) but require more refinement with respect to detailed pathway analysis as discussed below.
Functional Effects
Cardiac function
Modulation of cardiac function was one the first functional effects observed after administration of exogenous 3-T1AM (Table 2). Bradycardia was reported both in the conscious mouse and in the isolated perfused rat heart, while contractile performance was reduced in the working rat heart preparation (1,44). These effects were reversible after 3-T1AM washout or upon ADRβ stimulation by isoprenaline. The involvement of TAAR receptors was suggested by the observation that similar responses were induced by trace amines (IC50 = 109 μM for octopamine, 159 μM for β-PEA, and 242 μM for tryptamine) (46), and that many TAAR subtypes are expressed in rodent hearts. However, the role of Taar1 has been questioned on the basis of several considerations: in heterologous cell models, Taar1 stimulation is coupled to Gsα-stimulated cAMP production (1), which has opposite effects on cardiac function, namely increased contractility and heart rate; the relative potency of 3-T1AM and trace amines measured in the isolated rat heart was not consistent with the affinity for rat Taar1 (46); and recent evidence about the interaction of 3-T1AM with ADR (10) raises the possibility of alternative targets potentially involving Gi inhibitory signaling.
i.p., intraperitoneal; s.c., subcutaneous; b.w., body weight; i.c.v., intracerebroventricular; CNS, central nervous system.
The cellular signal transduction pathway appears to involve modulation in tyrosine phosphorylation of as yet unidentified target proteins, since contractile impairment was more severe in the presence of 37 μM concentrations of the tyrosine kinase inhibitor genistein, while it was partly rescued by the tyrosine phosphatase inhibitor vanadate (44). The final effector might be represented by the control of ionic homeostasis, since 3-T1AM treatment induced a decrease in systolic calcium transient, probably due to increased diastolic calcium leak from the sarcoplasmic reticulum, determining a depletion of sarcoplasmic reticulum calcium stores (45). Possible contribution by 3-T1AM mediated TRP signaling on calcium fluxes has not been analyzed in this context. Inhibition of a delayed rectifier potassium channel was also reported, which might be involved in the negative chronotropic effect. However, genistein, frequently alluded to as “specific tyrosine kinase inhibitor,” is also known to be an effective inhibitor both of TH binding to transthyretin and cellular TH transport and deiodination already at submicromolar concentrations (112 –114), apart from its interference with adenosine receptors at high 100 μM concentrations and competition for estradiol binding to its receptors at submicromolar concentration (115,116). Thus, genistein might also interfere with 3-T1AM availability, uptake, metabolism, and action at its target molecules.
The relevance of endogenous 3-T1AM in the control of cardiac function is uncertain. In rat cardiac homogenates, 3-T1AM concentrations were in the nanomolar range (3,117), while the EC50 for the cardiac effects of exogenous 3-T1AM was in the mid-micromolar range (44,46). This gap might be theoretically reduced if substantial compartmentalization exists for endogenous 3-T1AM, and/or substantial catabolism of exogenous 3-T1AM occurs before the receptors are reached. However, at present no experimental evidence is available to evaluate these issues.
On the other hand, surprising results were obtained when 3-T1AM was tested in an isolated rat heart model of ischemia-reperfusion injury (22). Pretreatment with 3-T1AM reduced irreversible ischemic injury. Because of the role of cytosolic calcium overload in ischemia-reperfusion, negative inotropic agents reducing cellular calcium availability are usually cardioprotective. In the case of 3-T1AM, however, protection was observed at concentration as low as 125 nM, which did not modify contractile performance, while higher concentrations did not affect ischemic injury. The transduction pathway also appeared to be different, since cardioprotection was abolished by the protein kinase C inhibitor chelerythrine and was not affected by genistein. It has been speculated that low 3-T1AM concentrations may trigger the transduction pathways involved in ischemic preconditioning, in which protein kinase C and mitochondrial effects are known to play a major role. At higher concentrations, this beneficial action might be overcome by different potentially adverse effects. Notably, in primary neuronal cultures cell viability was reduced after 24 hours of exposure to 50 μM 3-T1AM (23), concentrations much higher than those reported in blood. The pathophysiological implications of these findings, and/or their potential therapeutic exploitations, remain to be investigated. Recently, the activity of 3-T1AM metabolites has also been investigated, but TA1 did not produce any cardiovascular effect (28).
Thermoregulation
A single i.p. injection of 3-T1AM (50 mg/kg) led to a rapid, drastic, but transient decrease in rectal temperature in C57Bl/6 mice (1). Within 2 h, body temperature measured by rectal probe had fallen from 37°C to 29.5°C, and returned to normal body temperature 6–8 h after injection. The return to normal body temperature was also achieved by administration of β-adrenergic ligands, indicating a mechanism independent from classical catecholamine signaling. The mice did not show any signs of shivering, huddling, or piloerection but became inactive (1,23). A single 3-T1AM treatment (50 mg/kg) also significantly lowered body temperature in male IRC mice (lowest body temperature: 28.6°C after ∼1.4 h of treatment, and returned to the normothermic level after 5–6 h). A multi-dose 3-T1AM treatment (first i.p. injection and 2 s.c. 3-T1AM injections when body temperature returned to ∼32°C) was able to keep the animal in a prolonged hypothermic state (118). This hypothermia was confirmed in Djungarian hamsters (Phodopus sungorus) and probably occurs due to a decrease in metabolic rate (20). Interestingly, the extent of hypothermia was more pronounced in mice (∼6–7°C) than in long-photoperiod (∼2–3°C) and short-photoperiod hamsters (∼1°C). As these properties may have beneficial effects for emergency medical care (e.g., stroke therapy), major surgery, or organ transplantation, 3-T1AM is also a highly interesting molecule from a clinical perspective (119). So far, it was hypothesized that the trace-amine-associated receptor 1 (TAAR1) might be activated by 3-T1AM, mediating the effects via subsequent adenylyl cyclase activation. However, a recent report demonstrated that TAAR1 is unlikely to mediate the thermoregulatory response to 3-T1AM (95), as the hypothermic response to 3-T1AM administration was still maintained in TAAR1 knockout mice to a similar extent, and with the same dose response at 25 and 50 mg/kg body weight as in wild-type mice. Thus, the endogenous receptor(s) and underlying mechanisms causing the thermoregulatory effect of 3-T1AM are still elusive.
Furthermore, it is unknown if 3-T1AM metabolites might contribute to the profound thermoregulatory effects observed after 3-T1AM administration. Recently, it was shown that TA1, the main degradation product of 3-T1AM (27), had no effects on body temperature after a single TA1 injection (50 mg/kg, i.p.) measured by radio telemetry and repeated administration of TA1 (5 mg/kg, i.p. for 7 days) measured by rectal probe and infrared camera in male C57/Bl6 mice compared to sham-injected controls (28). Thus, it was concluded that TA1 does not contribute to the thermoregulatory effects observed after 3-T1AM administration in mice, suggesting that the oxidative deamination constitutes an important deactivation mechanism for 3-T1AM with possible implications for thermoregulatory functions (28).
Energy metabolism
3-T1AM received considerable attention following first reports on its rapidly occurring and marked hypothermic and metabolic effects (1,19,20,26,81,95,104,118), which were dose-dependent, reversible, and responsive to catecholamine administration distinct from other cryogens such 2-deoxy-D-glucose, mercaptoacetate, adenosine, 3′,5′-adenosine monophosphate (AMP) or hydrogen sulfide (H2S). Some of these 3-T1AM effects were initially interpreted in the context of hibernation or torpor, that is, metabolic states in free-living seasonal animals associated with markedly decreased food intake, energy expenditure, metabolic rate, respiratory quotient (RQ), and oxygen consumption, just the opposite of what is typically expected for an effect of a TH-derived metabolite. Comparable observations for dose-dependent pharmacological action of 3-T1AM on metabolism were made in several animal models such as rat, mouse, and the Djungarian hamster, which were explained by rerouting of metabolism from carbohydrate to lipid utilization. The outcome of these interventions manifested as weight loss mainly due to lipid mobilization and oxidation, supported by formation of ketone bodies and reflected by a switch of the insulin/glucagon ratio in the blood (1,20). Hypothermia might result from metabolic depression, impaired cardiac function, and altered neuronal regulation of peripheral functions. Species differences (more pronounced response in mice compared with the Djungarian hamsters) and seasonal differences became obvious (stronger hypothermia in long compared to short photoperiod hamsters) (20). On the other hand, 3-T1AM acted as an orexigenic agent both after i.p. injection of a lower dose (4 nmol/kg) in mice and after i.c.v. injection or direct administration to the hypothalamic arcuate nucleus in male rats. These bell-shaped dose-dependent effects were not accompanied by increased oxygen consumption (VO2) or locomotor activity after i.p. injection, but increased expression of the immediate early gene c-fos reflecting neuronal activation was found in the hypothalamus after direct arcuate nucleus 3-T1AM application (19). The authors provided some in vitro evidence that 3-T1AM might stimulate release of the orexigenic peptide NPY, but this has not yet been independently confirmed. A comparison between peripheral i.p. and central i.c.v. administration of different 3-T1AM doses in rats provided relevant mechanistic insight into regulation of glucose homeostasis by this biogenic amine (120). Already at low doses (0.5 mg/kg, i.c.v), 3-T1AM administration increased endogenous glucose production, blood glucose, glucagon, and corticosterone, while no or minor changes in insulin concentrations were observed, and central effects might prevail as underlying mechanism of 3-T1AM action. Also T0AM exerted similar effects and peripheral i.p. administration of the low 3-T1AM dose failed to mimic the effect of the i.c.v. application. Of interest, both 3-T1AM and T0AM i.p. administration decreased serum T4, T3, and, unexpectedly, thyrotropin concentrations (120), indicating either a direct thyroid effect and or major derangement of the pituitary–thyroid–periphery feedback axis by these TH metabolites. Whether this relates to elevated corticosterone concentrations reminding of acute stress interference on the hypothalamus–pituitary–thyroid axis or 3-T1AM-mediated activation of sympathetic and/or parasympathetic input on hepatic and pancreatic islet function needs more detailed and mechanistic animal experimental studies. To explain their observations, the authors referred to observations by Regard et al. (100) reporting on ADRα2a-mediated interference with Langerhans islet function activation, and to the effects of 3-T1AM and other TAM on plasma membrane and intracellular neurotransmitter uptake and transport by DAT, NET, and VMAT (95,105).
Experiments in at least one-year-old spontaneously overweight female CD-1 mice injected daily with a 10 mg/kg dose of 3-T1AM for eight days and analyzed for exhaled 13CO2 in breath by cavity ring down spectroscopy revealed increased lipolysis, with a significant weight loss persistent after discontinuation of 3-T1AM without changes in food consumption. Nuclear magnetic resonance spectroscopy of plasma indicated 3-T1AM stimulated lipolysis and protein breakdown (121).
At this point, cellular and intracellular targets and mechanisms contributing to altered energy metabolism, oxygen consumption via mitochondrial or other mechanisms (17,18) initiated by 3-T1AM exposure are still elusive and require analysis of cellular models, for example by Seahorse or related technologies, which may allow both analysis of oxygen consumption rate as well as extracellular acidification rate related to cytosolic glycolysis. No thermoregulatory or cardiac effects were observed after i.p. treatment of male mice with a single pharmacological TA1 dose (50 mg/kg, i.p.) or repeated application of TA1 in a lower dose (5 mg/kg, i.p., 7 days), indicating that this rapidly formed major metabolite of 3-T1AM, which exerts central nervous effects if applied locally, cannot provoke metabolic effects after peripheral administration (28).
Neuromodulation
The discovery of 3-T1AM was primed by the consideration that many amines derived from aromatic amino acid decarboxylation are chemical messengers (40). Trace amines and TAARs have been specifically regarded as involved in neuromodulation, that is, in regulating the response to neurotransmitters (122). 3-T1AM was originally detected in brain tissue (1), and i.c.v. administration of exogenous 3-T1AM induced significant functional responses, which have been extensively discussed elsewhere (24). Here, these effects will be summarized, and the potential interaction with established neurotransmitters will be discussed.
The most direct evidence for a neuromodulatory action is represented by electrophysiological recordings in rats showing that the rate of discharge of adrenergic neurons in the locus caeruleus was modulated by local 3-T1AM application (123). However, a note of caution must be introduced, since the EC50 for this action was 2.7 μM, which is probably significantly higher than endogenous 3-T1AM concentration.
In the mouse, i.c.v. injection of 3-T1AM produced complex effects on feeding behavior, depending on the dose and nutritional status. In mice fed ad libitum, food intake was increased by 1.2 nmol/kg 3-T1AM (19), while in fasting animals, a biphasic response was elicited, with 3.3 nmol/kg reducing food uptake and 51 nmol/kg producing the opposite effect (21). Modulation of other signaling pathways is suggested by the observation that the orexigenic effect was reproduced after local injection in the arcuate nucleus and was associated with the release of neuropeptide Y, a well-known orexigenic peptide (19). In addition, in fasting animals, the hyperphagia induced by high 3-T1AM doses was prevented by the monoamine oxidase inhibitor clorgyline, presumably through increased adrenergic drive (21).
In the mouse, a hyperalgesic effect was elicited by i.c.v. 3-T1AM, since the pain threshold measured by the hot-plate test was decreased (25). This effect has been attributed to TA1, produced by 3-T1AM oxidative deamination, and requires the integrity of the histaminergic system, since it was abolished by H1 histamine receptor antagonists, and it was absent in histidine decarboxylase knockout mice, which are severely depleted in histamine (2,29).
Histamine has also been involved in the prolearning and anti-amnestic effects that have been reported in a similar experimental model through the passive avoidance test, and are matched by increased curiosity, as revealed by the object recognition test (25). These effects, which might also be partly mediated by TA1, were abolished by H1 antagonists (2,29). Clorgyline depressed the anti-amnestic effects of 3-T1AM, suggesting a possible role also for the adrenergic system.
Other neurological actions have been reported for 3-T1AM, particularly modulation of the sleep–wake cycle with reduction in non-REM sleep (26). In this case, however, no hint was obtained with regard to the underlying mechanism and/or the involved neuromodulators.
Notably, in the papers addressing the effects on memory, learning, and pain threshold, whole-brain 3-T1AM and/or TA1 concentrations were measured, and it was observed that effective dosages increased tissue concentrations by about one order of magnitude over the baseline (25,29). The relatively small gap between endogenous levels and effective concentrations suggests that these effects might have physiological or pathophysiological relevance.
Preclinical observations
The initial discovery of combined hypothermic and metabolic effects ensued by existing observations on various behavioral and CNS actions of 3-T1AM (and its metabolites) significantly spurred research in this new area linking endocrinologists and neuroscientists at this interphase between TH and biogenic amines, as for example indicated by the priority research program THYROID TRANS ACT on “Translation of Thyroid Hormone Actions beyond Classical Concepts” funded by the German research organization DFG (
Such potent actions of 3-T1AM, its metabolites, and synthetic congeners are of eminent interest in emergency and critical care medicine, surgery, tissue transplantation, metabolic and eye clinics, as well as space science. Application of an endogenous biogenic cryogen derived from a hormone provides a rather safe and valuable “lead compound” to be tested and developed by the pharmaceutical industry for various medical applications (1,8,16,23,32,34,44,94,119,126). So far, only various small animal experimental paradigms and models have been studied and published, but no primate or human trials were initiated or published. To this end, more precise knowledge on site(s) of production, blood and tissue concentrations, target tissues and cells, cellular receptors, signal transduction cascades, effects, endpoints and biomarkers, pathways of inactivation and elimination, as well as dose–response studies for 3-T1AM in physiologically and pharmacologically relevant and validated animal models are required. While 3-T1AM represents a potent endogenous lead compound, its rapid metabolism indicates the urgent need for more stable, powerful, receptor-selective, and non-toxic agonistic and antagonistic analogs already available and partially tested in vitro and in vivo in rodents (13,111,127).
The most advanced areas in preclinical studies of 3-T1AM (and its metabolite TA1) are based on a number of experimental animal studies and include (i) its multifaceted cardiac effects (1,24,35), which are quite distinct and the opposite of classical T3 effects (ii) the metabolic function leading to increased lipid mobilization and oxidation (20,103,120,121,128); (iii) the pronounced dose-dependent hypothermic action (1,20,95); (iv) the alteration of glucoregulatory hormones of the pancreatic Langerhans islets (10,100,120); (v) the prevention of ischemia-associated insults such as stroke (22,23); and (vi) several promising 3-T1AM actions in the brain on behavior and sleep (2,19,25,26).
Open Questions and Pitfalls
The evidence collected since 2004 confirms that 3-T1AM is an endogenous substance, able to interact with specific receptors and producing significant functional effects (summarized in Table 2). On this basis, 3-T1AM is currently regarded as a biochemical messenger. However, the nature and the features of the putative signaling systems that involve 3-T1AM are not clear, and many crucial questions remain still open.
It is not known whether and where 3-T1AM is produced in peripheral tissues from circulating precursors, or whether it is rather synthesized at (a) specific site(s) and distributed systemically and thus qualifies as a bona fide hormone.
It is remarkable that among the broad class of amino acid–derived biogenic amines, 3-T1AM holds an exceptional position considering its unusually long half-life in human serum, probably due to its strong binding to apoB100, on one hand, and its distinct biosynthetic pathway, on the other hand. 3-T1AM is not stored and released from secretory vesicles in specialized cells like other biogenic amines and neurotransmitters—as far as currently known—but is probably generated from TH precursors. The observation that endogenous 3-T1AM has been found in several tissues after pre-analytical extraction and LC-MS/MS analysis by several but not all groups—while difficulties still prevail in determination of endogenous 3-T1AM concentrations in blood, tissues, or other body fluids such as CSF, as also reported in very recent LC-MS/MS-based publications (47,129,130)—raises the question of whether 3-T1AM might be considered rather as a para-or autocrine “tissue hormone” than as a classical endocrine signal, which is generated at (a) specific site(s) like the thyroid or the gastrointestinal mucosa, is transported via the blood stream bound to apoB100, and acts via receptor(s) at target cells, which react to this signal with altered function including generation of a feedback signal.
As a matter of fact, the modulation of 3-T1AM production and distribution is another crucial and still obscure issue, since the factors affecting its synthesis, transport, or catabolism have not been identified so far. Actually, there is still no direct evidence that 3-T1AM concentration in plasma or tissues may undergo developmental, phasic, or dynamic changes.
Many functional effects have been reported after administration of exogenous 3-T1AM in vitro or in vivo, but in most cases, the relevant receptors and transduction pathways have not been determined, and nor has it been possible to distinguish between physiological and pharmacological effects. The latter issue has been addressed by comparing endogenous 3-T1AM contents with the tissue concentration achieved after exogenous 3-T1AM administration. Tissue 3-T1AM levels were apparently in the nanomolar range, and some functional effects (particularly metabolic and neurological responses) were elicited at doses that induced an increase in tissue 3-T1AM of about one order of magnitude, suggesting that they might have physiological relevance. However, more direct evidence is required to support this conclusion, and it should be acknowledged that the concentrations that were determined in whole-tissue homogenates may not reflect the actual concentrations existing at the receptor level. The majority of studies addressing molecular targets and mechanisms of action of 3-T1AM and its major metabolites employed rather high micromolar concentrations in the in vitro models, and most of these studies did not address or mimic the crucial issue of the high affinity binding of 3-T1AM to apoB100, thus leaving a relevant gap of knowledge between in vitro and in vivo conditions.
While it was initially believed that 3-T1AM signaling represented a sort of negative feedback loop counteracting TH effects on thermoregulation and cardiac function, the present picture is much more complex, and it cannot be excluded that 3-T1AM production might contribute to some metabolic responses to the pleiotropic TH action. In the central nervous system, 3-T1AM and possibly TA1 have been suggested to act as powerful neuromodulators, which would be consistent with the role played in neurotransmission by other biogenic derivatives of aromatic amino acids, and might again contribute to the complex effects of TH on neural development, connectivity, and function.
The large number of open questions points to the need for technical breakthroughs. One of the most important is the development of a simple quantitative assay for 3-T1AM and derivatives, particularly in blood and other body fluids. As discussed previously, 3-T1AM can be detected by either mass spectrometry or immunoassay, but both techniques require substantial improvements and accurate validation. Large-scale assays of 3-T1AM in human blood might provide important information on the potential involvement of 3-T1AM or its derivatives in human disease, but such investigations could be misleading if the present technical problems and doubts are not solved.
Another breakthrough could ensue from the application of molecular biology techniques, such as the development of transgenic models of decreased/increased 3-T1AM production, as well as cell-specific receptor knockout or knockdown models. This will in turn depend on a better understanding of 3-T1AM metabolism and signal transduction. Novel aspects of 3-T1AM (patho-)physiology might be gained by a better knowledge of the endogenous distribution and biological function of various (conjugated) 3-T1AM metabolites shown to be formed in vitro (2,3,5,9,27,28,34,49,51). On the whole, many years after the first studies on TAM synthesis, the discovery of 3-T1AM has opened a novel research line that might provide significant advances in endocrinology. However, the complexity of the field and the technical problems of TAM research should not be overlooked, and great caution should be used before clinical implications are suggested. It should be kept in mind that more than 50 years elapsed between the discovery of thyroxine and the development of a reliable RIA assay for serum T4, which is not yet routinely analyzed by modern mass spectrometry in clinical practice, while 65 years were necessary to unravel the issue of T4 to T3 conversion. Hopefully, the pace of TAM research will be quicker, but premature conclusions might be misleading.
Footnotes
Acknowledgments
This work is supported by grants from the Deutsche Forschungsgemeinschaft within the DFG-SPP 1629 ThyroidTransAct to C.S.H. (HO 5096/2-1) and to J.K. ((KO 922/16-2,17-2), DFG-KFO 218 (KO 922/18-1)), and from Italian MIUR (PRIN08) and Fondazione Cassa di Risparmio di Lucca to R.Z.
Author Disclosure Statement
The authors have nothing to disclose.