
Abstract
Background:
Malignant thyroid teratomas are rare tumors with a poor prognosis. Little is known about their pathogenesis or treatment. Here, the case is reported of an adult woman with an aggressive thyroid teratoma with primitive neuroectodermal tumor (PNET) malignant transformation, successfully managed with neoadjuvant chemotherapy and surgery.
Patient Findings:
Sequencing of paired tumor and normal tissues revealed a DICER1 c.5438A>G (p.E1813G) somatic mutation in 56% of sequencing reads consistent with a driver event.
Summary and Conclusions:
To the authors' knowledge, DICER1 mutations have not been previously reported in teratomas but have been described in PNETs, suggesting a role in the malignant transformation of this case.
Introduction
T
Case Report
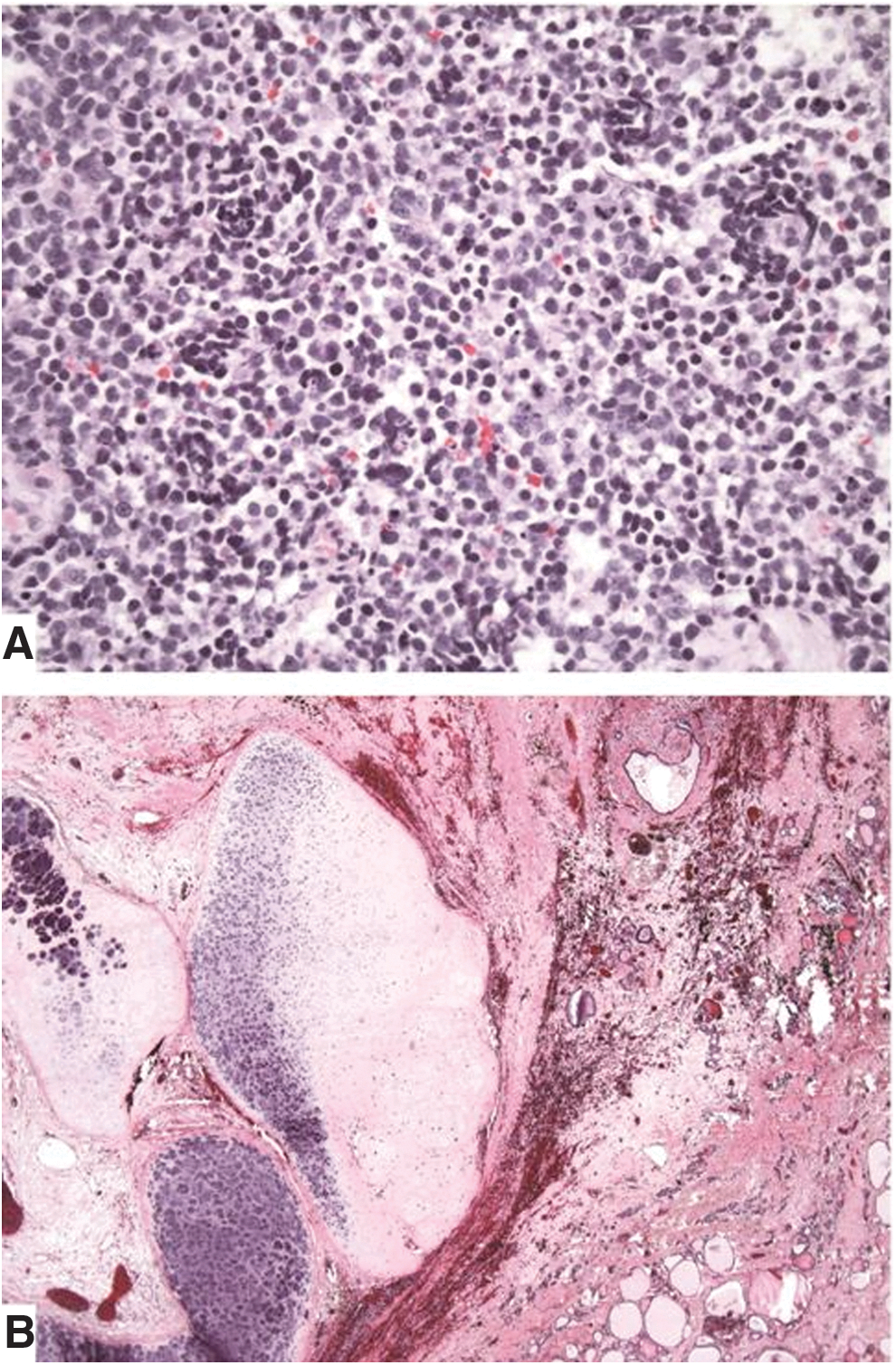
A 59-year-old African American woman presented to an outside hospital in February 2012 with a few weeks' history of rapidly progressing left neck swelling and hoarseness. Physical examination revealed a firm fixed 5 cm mass in the left thyroid region and diffuse firm fullness through the left levels II, III, and IV neck regions. Flexible nasopharyngolaryngoscopy examination demonstrated submucosal mass effect on the left side of the hypopharynx. Fine-needle aspiration of the left thyroid region mass was performed on February 25, 2012. Cytopathology identified a TTF-1-positive undifferentiated cancer. A computed tomography (CT) scan of the neck performed on the same day revealed a 3.7 cm × 3.6 cm × 6.7 cm heterogeneous soft-tissue density mass completely replacing the isthmus and left thyroid lobe with retrosternal extension into the anterior mediastinum (Fig. 1). A left lateral heterogeneous soft tissue mass of 2.7 cm × 2.8 cm × 2.1 cm in size in levels II, III, and IV, effacing the left piriform sinus, and multiple left jugular digastric lymph nodes with central necrosis measuring up to 1.6 cm in short axis were also seen. On March 5, 2012, she was seen at the authors' institution for further evaluation and recommendations. An open biopsy of the left level 2 mass was performed for additional testing on March 13, 2012. Pathology evaluation revealed a high-grade round-cell malignant neoplasm with focal rosette formation (Fig. 2A). The tumor cells were positive for SALL4 and TTF-1, and negative for CAM5.2, AE1/AE3, chromogranin, and synaptophysin.
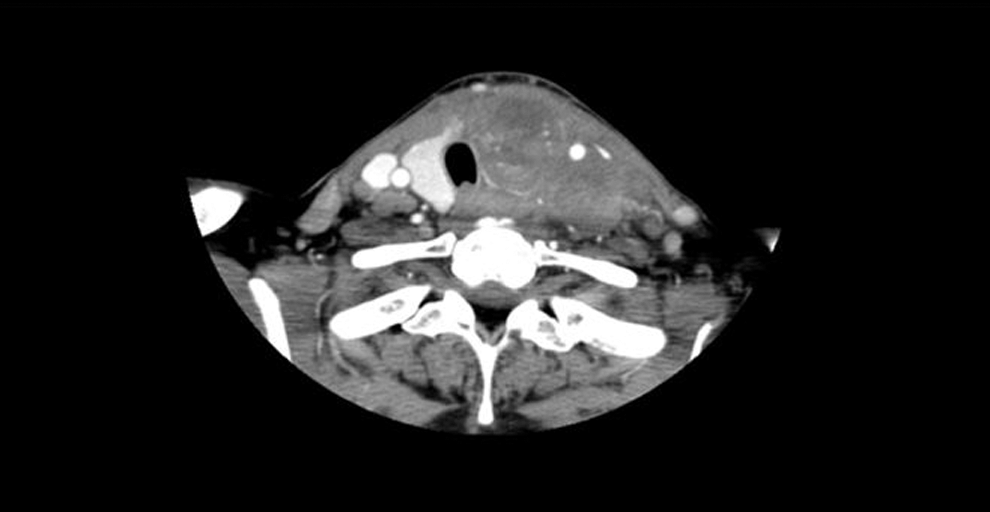
Neck computed tomography (CT) scan prior to chemotherapy demonstrates a large locally invasive mass centered in the left thyroid gland and involving multiple left lymph node levels.
(
Thus, the morphologic and immunophenotypic findings were consistent with a malignant neuroectodermal component of a teratoma. Interphase FISH studies were consistent with trisomy 12 in 58% of cells, but did not demonstrate the isochromosome 12p (i12p).
A few days post biopsy, she was admitted to the hospital with worsening neck swelling, pain, and shortness of breath. Neck CT confirmed disease progression with further left retrotracheal extension and tumor encasing the left carotid, as well as right paratracheal and right levels III and IV neck disease. She was then immediately started on cyclophosphamide, doxorubicin, and vincristine every three weeks, intercalating with ifosfomide and etoposide, PNET/Ewing sarcoma protocol, with a rapid symptomatic response. She underwent a total of four cycles of chemotherapy from March 2012 through May 2012, and achieved a clinical complete response. She tolerated therapy very well. A neck CT performed on June 5, 2012, revealed a 1.4 cm × 0.7 cm × 3.3 cm ill-defined hypodensity within the left thyroid lobe (Fig. 3). Surgery was then recommended for removal of what was thought to be the residual teratoma, which was delayed due to prolonged myelosuppression. Finally, on August 7, 2012, she underwent total thyroidectomy and neck dissection to encompass all initial sites of disease, including bilateral levels III–VI, and II on the left, and upper mediastinal node dissection, including chest nodal stations 4L (left lower paratracheal), 4R (right lower paratracheal), and 7 (subcarinal). The thyroidectomy specimen demonstrated residual teratoma of 3.5 cm in size involving the left lobe of the thyroid with extensive hyalinization, necrosis, dystrophic calcification, chronic inflammation, and pigment-laden histiocytes consistent with treatment effects (Fig. 2B). No residual high-grade round-cell malignant neoplasm was identified. The surgical resection margins were negative for tumor. The remaining neck and mediastinal specimens revealed histiocytic inflammation and lymphocyte depletion in multiple lymph nodes, consistent with treatment effect, with only rare atypical cells with degenerative changes. Thymic tissue revealed no significant pathologic change. She consented for oncopanel research testing, which is a multiplexed targeted assay that surveys exonic DNA sequences of 275 cancer genes to identify somatic mutations and copy number variations, in addition to 91 introns from 30 genes to detect rearrangements (5). Paired tumor and normal tissues were sequenced, identifying somatic mutations in DICER1 c.5438A>G (p.E1813G), TP53 c.743G>A (p.R248Q), TP53 c.376_splice (p.Y126_splice), and NF1 c.3160A>C (p.N1054H). No copy number alterations were observed involving the DICER1 gene. Notably, the DICER1 mutation was present in 56% of sequencing reads, consistent with a driver event, whereas the TP53 and NF1 mutations were present in 6–8% of reads, suggestive of subclonal tumor populations. The patient has been on active surveillance for more than four years since surgery, doing very well, without evidence of recurrent disease clinically or radiologically.
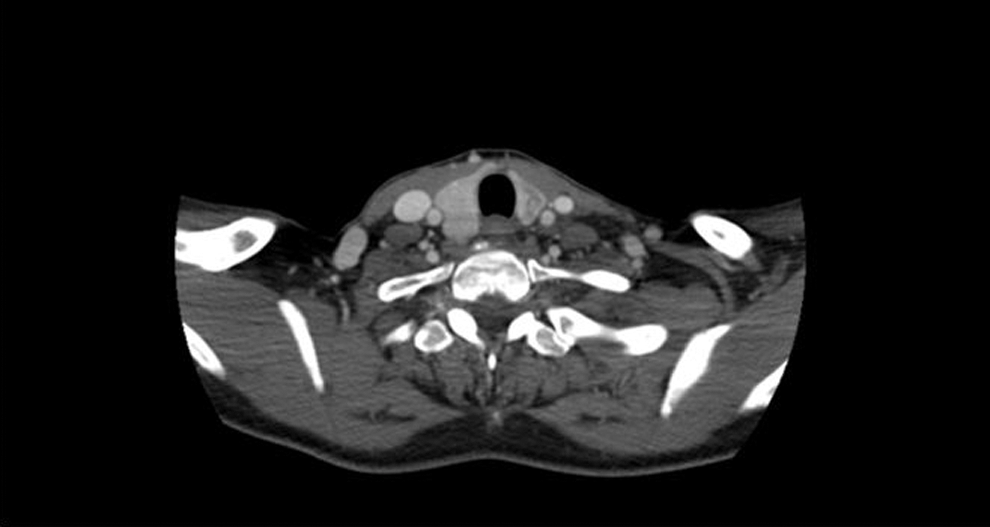
Neck CT scan after four cycles of chemotherapy demonstrates near complete response of the thyroid mass with a residual ill-defined hypodensity within the left thyroid lobe.
Discussion
Mature teratomas are relatively insensitive to radiation and chemotherapy, and surgery is considered the standard of care. In contrast, chemotherapy has been commonly used for patients with immature teratomas. Combinations of etoposide and cisplatin with bleomycin or vincristine have been commonly used prior to surgery in immature teratomas of the mediastinum with reported long-term survival (6). Although radiation therapy has been often applied in addition to surgery and chemotherapy, its benefit is unclear. For those with unresectable teratomas with malignant transformation limited to a single cell type, tailored chemotherapy directed toward that cell type has shown major responses and long-term survival in selected patients (7).
The present patient underwent four cycles of chemotherapy (PNET/Ewing sarcoma protocol, based on PNET malignant transformation), with complete pathologic response of the malignant component of her tumor. Surgery was then undertaken for removal of the residual, presumably mature component of the teratoma in the thyroid, as well as all sites of initial nodal disease in the bilateral neck and mediastinal areas. Oncopanel results revealed a somatic DICER1 mutation in 56% of sequencing reads. DICER1 is an endoribonuclease with two RNase III-like domains that plays a crucial role in the maturation of microRNAs, a group of noncoding small RNA molecules that regulate gene expression post-transcriptionally (8). Although DICER1 mutations have been associated with multiple malignancies, including thyroid neoplasia (multinodular goiter, adenomas, or differentiated thyroid gland neoplasia), and ovarian sex cord-stromal tumors (Sertoli–Leydig cell tumor, juvenile granulosa cell tumor, and gynandroblastoma), they have not been demonstrated in gonadal or extragonadal teratomas (9 –11). While many DICER1-associated tumors occur in the context of a germline mutation, there is a precedent for sporadic DICER1 mutation-driven tumors. Heravi-Moussavi et al. and Witkowski et al. demonstrated that >50% of ovarian Sertoli–Leydig cell tumors harbor monoallelic somatic mutations at one of the five hotspot sites (E1705, D1709, E1788, D1810, or E1813) in the metal-binding catalytic cleft of the DICER1 RNAse IIIB domain (11,12). Wang et al. demonstrated a dramatic reduction in expression of 5p-derived microRNAs in ovarian Sertoli–Leydig cell tumors carrying DICER1 hotspot mutations in comparison with those without, driving pseudodifferentiation of testicular elements and causing oncogenic transformation in the ovary (13).
De Kock et al. described three children carrying both germline and somatic DICER1 mutations, who developed differentiated thyroid cancer (9). The authors hypothesized that a second somatic hit was required in addition to a loss-of-function germline DICER1 mutation to initiate the thyroid carcinoma development. The targeted design of the sequencing assay employed in this study limited the ability to detect single nucleotide polymorphisms of known population frequency in and around DICER1. As a result, it was not possible to ascertain whether a copy-neutral loss of heterozygosity event may be serving as a “second hit” to the RNAse IIIb mutation. However, studies of Sertoli–Leydig cell tumors and other non-epithelial tumors of the ovary have similarly described somatic hotspot mutations in the RNAse IIIb domain of DICER1 without evidence of a second hit, and have ascribed a potential oncogenic role for DICER1 related to aberrant miRNA processing (12). The present patient's tumor had two TP53 and one NF1 gene missense mutations as subclonal variants. Although associations between these genes and teratomas have been previously reported (14 –16), it is unclear whether they have contributed to tumor development in this case. Finally, DICER1 mutations have been described in other PNETs, suggesting a potential role for the malignant transformation in this patient (17).
In conclusion, a rare case is presented of a woman with a malignant teratoma of the thyroid successfully managed with neoadjuvant chemotherapy and surgery. Despite previous reports suggesting the need for radiation therapy or a more prolonged chemotherapy course to optimize disease control (18), the added toxicity related to these may not translate in further benefit for the outcome of these patients. The identification of a DICER1 somatic mutation in this patient's tumor may have contributed to the PNET malignant transformation of the teratoma. The better understanding of the molecular changes of this rare disease may guide us on how best to approach these patients.
Footnotes
Author Disclosure Statement
No competing financial interests exist.