
Abstract
Background:
Anaplastic thyroid carcinoma (ATC) is a rare tumor, with poorly defined oncogenic molecular mechanisms and limited therapeutic options contributing to its poor prognosis. The aims of this retrospective study were to determine the frequency of anaplastic lymphoma kinase (ALK) translocations and to identify the mutational profile of ATC including TERT promoter mutations.
Methods and Materials:
One hundred and forty-four ATC cases were collected from 10 centers that are a part of the national French network for management of refractory thyroid tumors. Fluorescence in situ hybridization analysis for ALK rearrangement was performed on tissue microarrays. A panel of 50 genes using next-generation sequencing and TERT promoter mutations using Sanger sequencing were also screened.
Results:
Fluorescence in situ hybridization was interpretable for 90 (62.5%) cases. One (1.1%) case was positive for an ALK rearrangement with a borderline threshold (15% positive cells). Next-generation sequencing results were interpretable for 94 (65.3%) cases, and Sanger sequencing (TERT) for 98 (68.1%) cases. A total of 210 mutations (intronic and exonic) were identified. TP53 alterations were the most frequent (54.4%). Forty-three percent harbored a mutation in the (H-K-N)RAS genes, 13.8% a mutation in the BRAF gene (essentially p.V600E), 17% a PI3K-AKT pathway mutation, 6.4% both RAS and PI3K pathway mutations, and 4.3% both TP53 and PTEN mutations. Nearly 10% of the cases showed no mutations of the RAS, PI3K-AKT pathways, or TP53, with mutations of ALK, ATM, APC, CDKN2A, ERBB2, RET, or SMAD4, including mutations not yet described in thyroid tumors. Genes encoding potentially druggable targets included: mutations in the ATM gene in four (4.3%) cases, in ERBB2 in one (1.1%) case, in MET in one (1.1%) case, and in ALK in one (1.1%) case. A TERT promoter alteration was found in 53 (54.0%) cases, including 43 C228T and 10 C250T mutations. Three out of our cases did not harbor mutations in the panel of genes with therapeutic interest.
Conclusion:
This study confirms that ALK rearrangements in ATC are rare and that the mutational landscape of ATC is heterogeneous, with many genes implicated in the follicular epithelial cell dedifferentiation process. This may explain the limited effectiveness of targeted therapeutic options tested so far.
Introduction
A
ATCs originate from follicular cells and may derive de novo or from pre-existing well-differentiated thyroid cancers (WDTCs) such as papillary thyroid carcinomas (PTCs), follicular thyroid carcinomas (FTCs), or poorly differentiated thyroid carcinomas (PDTCs) by accumulation of various genetic alterations (4 –9).
Numerous genetics studies have explored the ATC mutational landscape: 90% of ATCs harbor a mutation in the MAP-kinase pathway, BRAF is altered in 25–91% (10 –13) and RAS in 6–50% (13 –18). Gain of function mutations in the PIK3CA gene and amplification of the PIK3CA genomic locus are found in 18–40% and in about 40%, respectively, suggesting that alterations in the PI3K-dependent pathway play a relevant role in the pathogenesis of ATCs (13,15,16,18 –20). Between 25% and 60% of ATCs also harbor mutations in the CTNNB1 gene that activates the WNT pathway (21). Furthermore, loss of function alterations of tumor suppression genes such as PTEN, which inhibits the activation of the PI3K pathway, have been reported in 4–16% of ATC (15,16,18). TP53 is another gene commonly mutated/inactivated with a frequency of 12–83%, causing inactivation of apoptosis and cell-cycle progression.
The therapeutic strategy for ATCs is based on a multimodal approach (surgery, radiation, chemotherapy) for operable disease. Patients with advanced/metastatic disease may be eligible for radiochemotherapy, but most patients are considered for palliative care (5) or clinical trials. Multi-targeted receptor tyrosine kinase inhibitor or angiogenesis inhibitor therapy showed no significant response (22 –25). However, few case reports have shown tumor responses with targeted therapy: a spectacular response to vemurafenib was seen in a 51-year-old man with BRAF-mutated ATC (26). An 18-month response was described in a patient with metastatic ATC (27) treated with everolimus, an inhibitor of the mammalian target of rapamycin (mTOR), until resistance developed. Finally, the authors' team recently reported a remarkable response to crizotinib in a 71-year-old woman with anaplastic lymphoma kinase (ALK)-rearranged ATC (28).
Although the physiological function of ALK is still poorly characterized, it is known that rearrangement of the ALK gene in a tumor leads to constitutive activation of tyrosine kinase activity of ALK-fusion chimeric proteins and several key downstream cellular pathways (29). ALK rearrangements have been described in many neoplasms (30). In ALK-rearranged non-small-cell lung carcinomas, crizotinib, a tyrosine kinase inhibitor of MET, ALK, and ROS1, is now a new therapeutic standard. In thyroid cancers, the authors' team and others have described a novel ALK rearrangement with the Striatin gene (STRN), but its exact frequency in a wider population is not well known (31,32). Due to the growing number of ALK-driven cancers, its limited expression in adult normal tissues, and the existence of efficient ALK inhibitor (33), ALK appears to be a promising oncogene to target for personalized therapy in thyroid carcinomas (34).
Another oncogenic event in thyroid carcinomas has emerged. Mutations in the promoter region of the telomerase reverse transcriptase (TERT) have recently been described in different types of cancer, including follicular cell-derived thyroid cancer (35). TERT promoter mutations were screened in six studies including 252 cases of ATC. One hundred (39.7%) tumors harbored TERT promoter mutations, and this prevalence seems to increase with the dedifferentiation process (36). TERT plays a dominant role in the activation of telomerase during malignant transformation of cells and can also modulate the expression of growth-controlling genes that regulate cell proliferation (37) and may participate in oncogenesis.
This study collected a large series of 144 ATCs from 10 participating centers of the national French network for management of Refractory Thyroid Tumors (TUTHYREF). They were screened for ALK rearrangements using fluorescence in situ hybridization (FISH) to establish the exact frequency of this alteration in this tumor type, and for mutational alteration by next-generation sequencing (NGS) in order to identify potential driver mutations. Subsequently, Sanger sequencing was performed for TERT promoter mutations.
The aims of the study were to understand ATC carcinogenesis better and to identify new targeted therapies considering the failure of conventional therapies and the heterogeneity of responses in trials with targeted therapies.
Materials and Methods
Population and tumor sample acquisition
In this retrospective study, tumor samples from 1973 to 2014 were collected from 10 participating centers of the TUTHYREF. The 144 patients included 86 (59.7%) women and 58 (40.3%) men, with a mean age of 68.3 years at diagnosis (range 38–94 years). All the samples were fixed in buffered formalin, except for 23 cases that were fixed in acetic formalin and three cases in Holland Bouin fixative. All samples and techniques were centralized in one center (Institut Bergonié, Bordeaux, France).
Histological features
Diagnosis and subtype classification were performed according to the World Health Organization classification in Pathology and Genetics of Tumours of Endocrine Organs (9). When present, differentiated components were classified into FTC or PTC for well-differentiated components and poorly differentiated components were defined using criteria described in the Turin algorithm (38).
Tissue microarray construction
Tissue microarrays (TMA) were designed from 107 blocks out of the 144 collected samples. The rest of the samples (37 cases) contained insufficient amounts of tissue. For each case, the anaplastic component was identified on a hematoxylin–eosin–saffron (HES) staining slide. Tissue cores, 0.6 mm in diameter, were removed from fixed paraffin-embedded tissue blocks and arrayed on a recipient paraffin block using a tissue arrayer (Beecher Instruments, Sun Prairie, WI). Each component was punched in triplicate. Sections of the array (5 μm thick) were cut and placed on glass slides. An independent TMA incorporating all cases not fixed in buffer formalin was designed, and an ALK-positive lung tumor was incorporated as an external positive control for FISH.
FISH
The FISH assay for the ALK gene was performed using the DAKO Histology FISH Accessory Kit (Agilent Technologies, Santa Clara, CA), as previously described, according to the manufacturer's instructions. Interphase molecular cytogenetic studies using a commercially available ALK probe (Vysis LSI ALK Dual Color, Break Apart Rearrangement Probe; Abbott Molecular, Abbott Park, IL) was performed on a 4 μm paraffin-embedded section cut from either TMA blocks or whole section blocks (for the 37 cases with insufficient material). Nuclei were scored for non-rearranged patterns (red and green fusion signals) and rearranged and unbalanced patterns (split of red and green signals or extra single red signals) using a Nikon Eclipse 80i fluorescent microscope with appropriate filters. The positive cutoff chosen was the same used in the lung sample (≥15%). Pictures were captured using a Hamamatsu C4742-95 CCD camera and analyzed with the Genikon software (ALPHELYS Lab Technologies, Plaisir, France).
NGS
All samples with enough material were screened by NGS. The tumor sample selected contained at least 50% neoplastic cells. DNA was extracted with the QIAamp DNA FFPE Tissue Kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions and was quantified with the Qubit kit (Qubit™ dsDNA BR Assay Kit; Molecular Probes, Inc., Eugene, OR). The Ion Ampliseq cancer panel v2 (Ion Torrent; Life Technologies, Foster City, CA) encompasses hotspot mutation regions of the following genes: ABL1, AKT1, ALK, APC, ATM, BRAF, CDH1, CDKN2A, CSF1R, CTNNB1, EGFR, ERBB2, ERBB4, EZH2, FBXW7, FGFR1, FGFR2, FGFR3, FLT3, GNA11, GNAQ, GNAS, HNF1A, HRAS, IDH1, IDH2, JAK2, JAK3, KDR, KIT, KRAS, MET, MPL, MLH1, NPM1, NOTCH1, NRAS, PDGFRα, PIK3CA, PTEN, PTPN11, RB1, RET, SMAD4, SMARCB1, SMO, SRC, STK11, TP53, and VHL. Target amplicon libraries were generated with the Ion Ampliseq kit v2.0, and amplicons were ligated to adapters with the Ion Xpress barcoded adapters 1–96 kit. Libraries were quantified with the Qubit dsDNA HS Assay Kit. Multiplexed barcoded libraries were amplified by emulsion polymerase chain reaction (PCR) on ion sphere particles, and sequenced on a ion 318 chip V2 with a PGM sequencer using the Ion Ampliseq PGM sequencing according to the manufacturer instructions. Torrent suite software v4.4.2 was used to parse barcoded libraries and align reads to the reference genome hg19. Variants were identified with the Torrent Variant caller software v4.4.2.1 with the following parameters: somatic low stringency custom (SNP threshold detection 2% and indel 5%).
Sanger sequencing for TERT promoter mutations
The same DNA samples as for NGS were used. A fragment of the TERT promoter, containing the 1,295,228C>T and 1,295,250C>T loci corresponding to nucleotides -124 G > A and -146 G > A upstream of the initiation codon ATG, was amplified by PCR using the following primers: 5′-CAC CCG TCC TGC CCC TTC ACC TT-3′ (forward) and 5′-GGC TTC CCA CGT GCG CAG CAG GA-3′ (reverse). The PCR condition included an initial denaturation step at 95°C for 10 min, 40 cycles of 95°C denaturation for 30 s, 55°C annealing for 45 s, 72°C elongation for 45 s, and a final elongation step at 72°C for 10 min. The quality of the PCR products was analyzed by 2% agarose gel electrophoresis. The PCR products were sequenced using a Big Dye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA) on a 3130XL Genetic Analyzer (Applied Biosystems) to identify the mutation.
Results
Histological features
Anaplastic components were subclassified into histological variants: giant cell (four cases); spindle-shaped cell (34 cases); epidermoid cell (14 cases); pleomorphic cell (55 cases); pleomorphic and epidermoid cell (two cases); pleomorphic and giant cell (eight cases); spindle-shaped and giant cell (three cases); spindle-shaped and epidermoid cell (one case); spindle-shaped and pleomorphic cell (15 cases); spindle-shaped, pleomorphic, and giant cell (three cases); and paucicellular (five cases).
Among these cases, a differentiated component was identified in 31 cases: 21 with well-differentiated (19 PTCs and two FTCs), eight with poorly differentiated and two cases with well- (FTC) and poorly differentiated components.
FISH
It was possible to analyze 90 (62.5%) anaplastic cases. None of the samples fixed in Holland Bouin fixative and 19/23 samples in acetic formalin fixative were interpretable. Only one (1.1%) case was positive with a threshold percentage considered as borderline (15%; Fig. 1A). This case (Fig. 1B) was associated with a well-differentiated PTC component (Fig. 1E). An ALK immunohistochemistry assay performed with the D5F3 antibody clone found a negative anaplastic component (Fig. 1C) and a “1+/doubtful” positive well-differentiated component (Fig. 1F) furthermore “negative” for FISH (Fig. 1D).
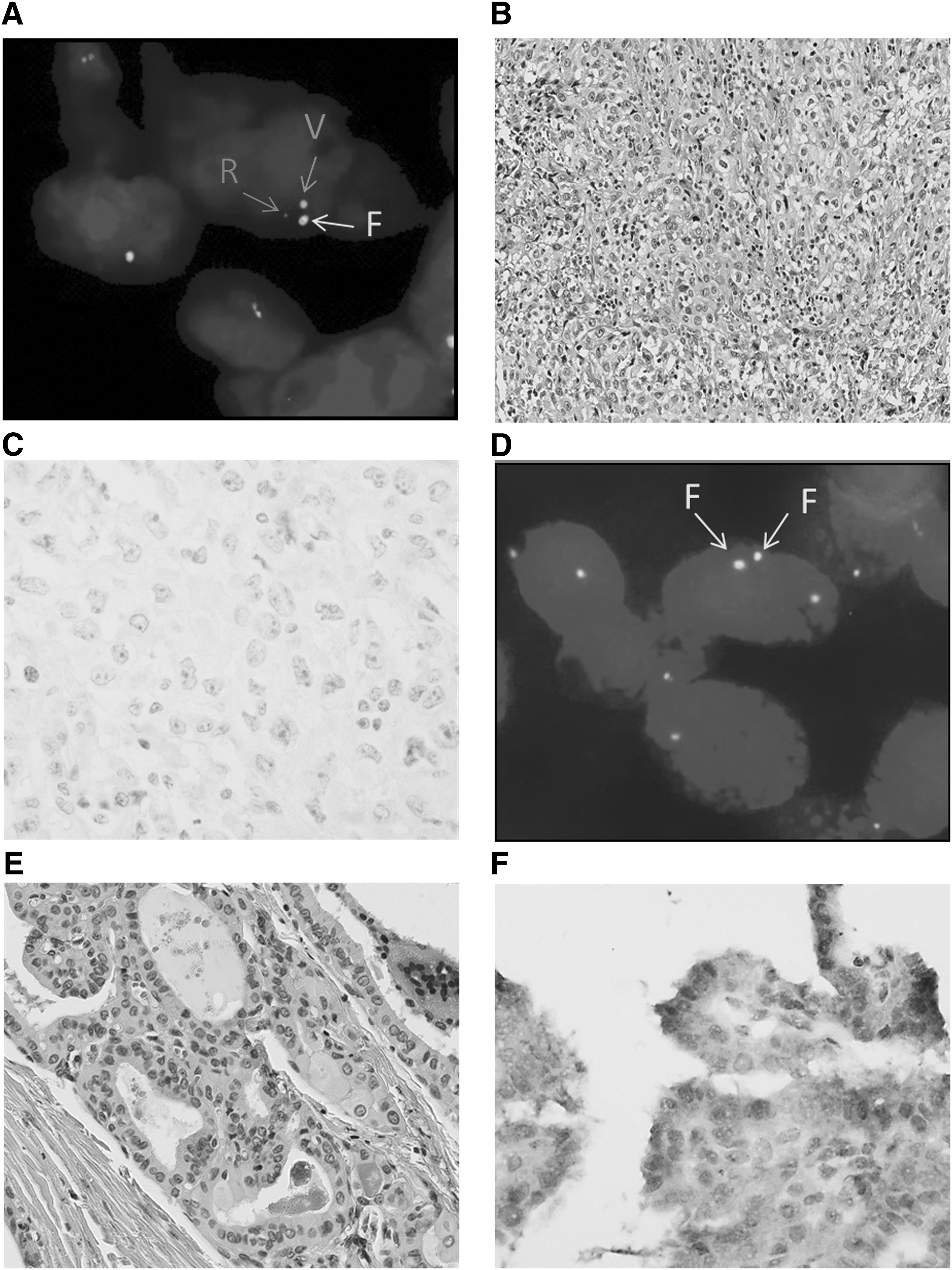
Case 136 (hematoxylin–eosin–saffron staining [HES], immunohistochemistry [IHC], and fluorescence in situ hybridization [FISH]). (
NGS sequencing
It was possible to analyze 94/144 (65.3%) cases with NGS, including 16 cases with an alternative fixative other than buffered formalin. Nine cases had very little material for DNA extraction, and 41 cases were not analyzed due to the lack of DNA amplification. The mean number of reads in analyzed cases was 468,490, with 95% of reads on target. The mean depth was 1974X and the average homogeneity of regions of interest (% bases on target by at least 20% of the average depth) was 93.4%.
Of the 135 cases with already extracted DNA, 111 were investigated with Sanger sequencing. Thirteen cases had no DNA left after the NGS technique. Twenty-four cases were not analyzed due to the lack of DNA amplification (all these cases were also not analyzable with the NGS technique). Of 144 cases, 98 (68.1%) were finally amenable to analysis.
A total of 152 exonic non-synonymous variants and 58 intronic variants (including 53 TERT promoter mutations), totaling 210 mutations, were identified (Table 1). The number of somatic variants per tumor ranged from one to five in 51 tested genes, with a mean number of total and non-synonymous variants of 2.14 per tumor. An overview of the results by sample is shown in Table 2, and a detailed list of the sequencing data for all the samples is included in Supplementary Table S1 (Supplementary Data are available online at
Three cases with two TP53 mutations.
One case with two ERBB2 mutations.

The most frequent alteration was found in the TP53 gene: 54 different mutations, including three cases with two TP53 mutations out of 144 (54.4%) cases.
Overall, the MAP-kinase pathway (H-K-N RAS and BRAF) was found altered in 53 (56.4%) cases, with the most frequent alteration being in the RAS genes (H, K, or N-RAS) with a total of 40 (42.6%) mutations. The main mutation was found at codon 61 of NRAS (100% of the 28 cases with NRAS mutation), followed by HRAS (six at codon 61 and two at codon 13) and KRAS (two at codon 12 and two at codon 61). BRAF was altered in 13 (13.8%) cases in exon 15. Twelve were the classical p.V600E mutation, and the remaining one was a rare mutation also located in exon 15 p.N581S. BRAF mutations were associated in 2/13 (15.4%) cases with an altered PI3K/AKT/PTEN pathway, and in 3/13 (23.1%) cases with a TP53 mutation. The p.N681S BRAF mutation was associated with a NRAS mutation and a TERT promoter mutation. Finally, after TERT promoter sequencing, BRAF was the only driver mutation in 2/13 (15.4%) cases.
A PI3K/AKT/PTEN pathway alteration defined by either a PIK3CA or AKT1 or PTEN mutation was found in 16/94 (17.0%) cases. Six non-synonymous PIK3CA mutations (five at codon 1047 and one at codon 545) were identified, as well as two AKT1 mutations in exon 3 p.E17K. No other mutations were observed in 3/16 (18.8%) cases, an association with a BRAF mutation in 2/16 (12.5%) cases, a RAS alteration in 4/16 (25.0%) cases, a TP53 mutation in 9/16 (56.3%) cases, and a TERT mutation in six (37.5%) cases.
Eight (8.5%) PTEN mutations were found in the cohort: two (25.0%) were unique, four were associated with a TP53 mutation (50%), two with a TERT promoter mutation (25%), and one (12.5%) with a BRAF mutation.
Finally, only 16/94 (17.0%) cases had an isolated mutation of the MAP-kinase pathway or of the PI3K/AKT/PTEN pathway. Overall, one of these two pathways was affected in the majority (62/94; 66.0%) of cases.
Fifty-three (54.0%) mutated cases were found for one of the two hotspots described in the literature in the TERT promoter: 43 cases for -124G>A (C228T) and six cases for -146G>A (C250T). No previously unknown mutation was identified. Among these cases, TERT promoter mutations were the only mutations in four (4.25%) cases analyzed by NGS, associated with RAS mutations in 19 (20.2%) cases and BRAF alterations in nine (9.6%) cases.
The other genes of the panel were rarely affected: four (4.3%) mutations in ATM, two (1.1%) in a unique case in ERBB2, one (1.1%) in MET, and one (1.1%) in ALK.
Only three cases did not exhibit any somatic mutations in the 51 genes tested. The mean depth for these cases was 1685X [1208;1993]. Macrophage abundance in this sample was screened after HES staining and with immunohistochemistry with CD163. Macrophage infiltration was scored between 0 and 3. The mean score for these cases was 2.5. Finally, no mutation has been detected in the cohort in the following genes of the panel: CDH1, CSFR1, CTNNB1, EGFR, EZH2, FGFR2, FGFR3, FLT3, GNA11, GNAQ, HNF1A, IDH1, IDH2, JAK2, KIT, MPL, MLH1, NOTCH1, NPM1, PDGFRα, PTPN11, and SRC.
Discussion
ALK rearrangements in ATC
ALK rearrangements in thyroid carcinomas were first described in a series of PTC (31) followed by ALK point mutations in ATCs (11.1% prevalence) (39). A new translocation of the ALK gene with the Striatin gene (STRN) was recently identified in a 71-year-old woman with an ATC derived from PTC who was successfully treated with crizotinib (28). Despite the series being the largest ATC series analyzed for ALK rearrangements, only one positive case was found by FISH. The rearrangement rate was equal to the threshold limit (15%), so the estimated prevalence in the series was low and only 1.1%. This could be explained by the fact that ALK rearrangements in thyroid cancer seem to be linked to PTC origin and, in the series, the rate of ATC with an obvious PTC component is low (19/144; 13.2%). However, it is also possible that cryptic rearrangements of the ALK gene do exist but are not detectable by FISH and may require a high-throughput technique such as RNA sequencing for detection. Of note, similar to these results, two recent clinicopathologic studies showed the absence of ALK translocations in 20 ATC cases analyzed by FISH (40) and 33 cases characterized by transcriptomic NGS analysis (13). In contrast, the later study found three cases with ALK translocations (with STRN, EML4, and a newly identified fusion with CCDC149) in poorly differentiated samples. This could also be explained by the fact that thyroid cancers driven by fusion genes are somewhat less virulent and may less often progress to ATC.
Mutational landscape of ATC by NGS
In the present large cohort of ATC patients, a NGS approach was chosen for 50 genes of oncogenic and therapeutic interest. The study was completed with TERT promoter Sanger sequencing, an emerging oncogenic event in thyroid carcinomas not included in the NGS panel.
The third most frequent alteration in the series (after TP3 and TERT promoter mutations) was in the RAS proteins, with a total of 40 (42.6%) mutations. These mutations are frequent in a variety of thyroid tumors from benign follicular adenomas (20–25%) to aggressive ATCs (20–30%), suggesting a role in early tumorigenesis (34). Since RAS mutations are present in thyroid adenomas, this anomaly seems insufficient in itself to explain the dedifferentiation process and the existence of ATC cases with an isolated RAS mutation. Nevertheless, it also represents an important molecular alteration in poorly differentiated carcinomas (41). NGS analysis of 84 poorly differentiated carcinomas by Fagin et al. showed that RAS mutations were found especially in PDTCs fulfilling the Turin definition (38), the criteria that was used to classify the PDTC components in this study.
BRAF mutations were found in 13.8% of cases. All mutations were p.V600E and were mutually exclusive to (K-H-N)-RAS mutations, except one p.N581S that had coexistent NRAS and TERT promoter mutations. Moreover, two of these cases harbored a coexistent PIK3CA mutation. In two cases, the BRAF mutation was the only detected genetic alteration. Despite a well-known correlation between the presence of a BRAF mutation and tumor aggressiveness in PTCs (42,43), BRAF mutations are less frequent in ATCs (as in the present cohort) than in PTCs (prevalence of 45–50%) (44). Since BRAF mutations do not appear to occur during tumor dedifferentiation but are relatively early and frequent events in PTC carcinogenesis (45), not all ATCs may be derived from PTCs. Therefore, BRAF mutations seem to be the most prevalent mutation in PDTCs, defined based on MSKCC criteria (46), that is, high mitotic rate and necrosis, irrespective of growth pattern (13). This could be a part of a lesional continuum driven by BRAF alterations. Moreover, case reports of ATCs with BRAF mutations show interesting therapeutic responses to BRAF inhibitors, albeit partial or temporary (26,47). De novo resistance of BRAFV600E -mutated cancer cells to vemurafenib has been attributed to the rapid rebound of MAP-kinase/ERK signaling. Indeed, BRAF-mutant cells have been reported to be MEK-dependent and selectively more sensitive to MEK inhibition than either RAS-mutant or both BRAF and RAS wild-type cells. As MEK is a key downstream signaling mediator of the MAP-kinase pathway, the use of co-inhibitors has been proposed in anticancer therapies (48). A clinical trial using dabrafenib (a BRAF inhibitor) and trametinib (a MEK inhibitor) in ATC is currently ongoing (49).
PI3K/AKT pathway mutations (especially PIK3CA mutations) have been identified in recurrent and metastatic tumors and only in thyroid lesions other than PTCs (50). Hence, a study focused on PIK3CA mutations in ATC and its various components found an increased prevalence of PIK3CA mutations in the poorly differentiated components compared with well-differentiated components of the same lesion (18). The prevalence of PIK3/AKT pathway alteration in the present series was lower than that reported in the literature for ATCs (15–25%) and was close to that found for follicular carcinomas (41). Prior studies on ATCs found PIK3CA mutations/copy number gains to be overlapping with the presence of BRAF or RAS mutations, or increased p53 expression (51), suggesting that PIK3CA alterations often cooperate with other oncogenic events in these types of tumors.
In general, the frequent coexistence of mutually exclusive mutations is further evidence for the mostly heterogeneous genetic background of ATCs, and it can be explained either by genetic instability leading to passenger mutations or by clonal heterogeneity within the tumor. For example, 6/94 (6.4%) cases had a co-alteration affecting the MAP-kinase (including BRAF) and PI3K/AKT pathways. The presence of a RAS mutation is often associated with resistance to selective inhibition of the MAP-kinase or PI3K pathways (52).
Two cases in the present ATC cohort had an already reported (18) coexistent mutation of RAS and PIK3CA (2.1%) as well as a BRAF mutation (53). Coexistence of a BRAF mutation that alters and activates the PI3K/AKT pathway has also been recently reported with a prevalence of 5.5–9% for BRAF-PIK3CA, mostly in aggressive cancers such as recurrent/metastatic cancers, PDTCs, and ATCs (51,54,55). However, such mutations can widely coexist, leading to PI3K/AKT pathway activation in up to 81% ATC cases according to the literature (10,18,53,56). In summary, 66.0% (n = 62) of cases had a direct alteration of one of the genes involved in the MAP kinase or PIK3/AKT pathway.
Tumor suppressor gene alterations are also prevalent in ATCs. TP53 tumor suppressor gene mutations are the most frequent anomaly in the present cohort and in ATC in general. TP53 mutations are present in about half of all the cases of the cohort (n = 51; 54.4%). The prevalence rate in the literature ranges from 48% (56) to up to 70–80% (57,58). In contrast to ATC, TP53 mutations are rarely encountered in differentiated carcinomas. TP53 damage in a differentiated carcinoma is likely a key factor of dedifferentiation and major chromosomal instability (59,60).
PTEN mutations (n = 8; 8.5%) occur at a slightly lower rate to that found in the literature, with a prevalence of around 10–20% (41). PTEN gene alterations (point mutations, deletions) are infrequent in sporadic thyroid tumors. It also seems that PTEN function is frequently altered in ATC but that PTEN mutations are not necessarily the predominant mechanism, arguing in favor of a late event in thyroid tumorigenesis (15). The discovery of PTEN methylation in thyroid tumors (61) could also explain its loss of function. Hypermethylation of PTEN plays a role in the negative regulation and activation of the PI3K-AKT pathway, and this epigenetic modification may be secondary to PI3K-AKT pathway alterations (62).
Four cases in the cohort had a PTEN/TP53 co-mutation. Among them, one had a supplementary TERT promoter alteration. Experiments in ATC mouse models with this double alteration show that despite the simultaneous loss of PTEN and p53, tumor development is not remarkable, suggesting that additional genetic alterations may be needed (63).
Two recent reviews have evaluated the importance of TERT promoter mutations in thyroid cancers (36,64). The prevalence seems to increase with the dedifferentiation process and ranges from 10.4% to 11.3% in PTC, from 17.1% to 17.3% in FTC, from 40.5% to 43.3% in PDTC, and from 39.7% to 40.1% in ATC (36,64). Two hot spots have been described with no overlap: C228T and C250T, the former far more prevalent than the latter (36). Such mutations confer enhanced TERT promoter activity by generating a consensus binding site (GGAA) for E-twenty six (ETS) transcription factors within the TERT promoter region (35). Biologically, TERT is the catalytic protein subunit of telomerase, whose function is to add telomeres at the end of chromosomes maintaining chromosomal integrity and genome stability. Normally repressed in human somatic cells, TERT activity is expressed in several cancer cells of various types (65 –67). However, TERT also has a telomerase-independent role. In fact, in vitro experiments have demonstrated that the overexpression of TERT, even when it lacks telomerase activity, significantly elevates the cell proliferation rate without enhancing telomerase length (68).
Furthermore, consensus binding sites for ETS transcription factors in the TERT promoter lead to the overexpression of TERT and can be upregulated by a synergistic BRAF co-alteration (69). This phenomenon is thought to occur with RAS alterations by activating the PI3K pathway. Clinically, in a large study, Xing et al. demonstrated that coexistence of the BRAFV600E and TERT promoter mutations is associated with particularly aggressive tumors and poor clinical outcomes of PTC (70). TERT promoter mutations, as well as the BRAFV600E mutation, are generally associated with aggressive thyroid tumor characteristics, tumor recurrence, and patient mortality (36). In the present cohort, nine cases had a TERT promoter mutation associated with a BRAF mutation, and 19 cases were found to have a coexistent RAS alteration.
Some genes, rarely described in thyroid carcinogenesis, have emerged in this study such as a not previously reported MET mutation p.I166T (c.497T>C). In contrast to MET amplification reported in approximately 12% of ATCs (41), MET mutations in thyroid cancers have not been reported. Similarly, a single not previously reported mutation was found in exon 23 of the ALK gene p.D1203H (c.3607G>C) associated with an APC p.G1317Q (c.3949G>C) mutation in exon 16. These two alterations could be targeted for therapy based on functional studies (71). One of the cases had two not previously reported mutations, p.D873N (c.2617G>A) and p.A763T (c.2287G>A), in two different exons (19 and 21) in the region encoding the ERBB2 tyrosine kinase domain. Studies on ERBB2 in thyroid tumors, especially in ATCs are rare. An association between ERBB2 overexpression and tumor aggressiveness in differentiated thyroid carcinomas have already been reported (72 –74). ERBB2 mutations could be a potential target for therapy with an ERBB2 anti-tyrosine kinase whose effectiveness has been demonstrated in vitro (75 –77).
RET mutations are mostly described in medullary thyroid carcinoma (78,79). A single RET mutation has been identified in a series of 22 ATCs (56), associated with TP53, CTNNB1, and RASAL1 mutations. In the present cohort, two RET mutations, one associated with TP53 p.S653C (c.1958C>G) in exon 11 and the other with SMAD4 p.G252R (c.757G>A), not referenced in the COSMIC database and 2 ERBB2 mutations [p.A763T (c.2287G>A), p.D873N (c.2617G>A)] were present. Both of them were associated with a TERT promoter mutation.
SMAD4 mutations (one case in the present cohort) have been described in 15/56 (27%) thyroid lesions, both benign and malignant (80).
Finally, one case in the present series had a single frame-shift mutation in STK11 (p.P281Rfs*6) associated with PTEN and TP53 mutations. Two STK11 mutations have already been described recently in ATCs (13). Everolimus is an FDA-approved oral allosteric inhibitor of mTOR. Inactivating mutations in the tumor suppressor genes TSC1, TSC2, and STK11 result in mTOR-pathway activation and could be targeted by mTOR inhibitors (27).
Finally, in three cases, no mutation in the genes covered by the panel was identified. A lack of mutation in oncogenes or tumor suppressor genes has been previously reported in an extensive whole-exome sequencing study (56) and could be explained by (i) other mechanisms such as other known translocations involved in these tumors or epigenetics mechanisms, or (ii) mutations in genes not included in the initial NGS panel. The complementary study with TERT promoter Sanger sequencing was able to detect four mutations in cases with no mutation in 50 NGS panel genes. The absence of mutations could not be explained by a putative macrophagic infiltration, as the cellularity of the extracted areas was at least 50%, far above the sensitivity of the NGS technique and the average sequencing depth was 1685X for the three cases without mutation.
Further molecular analyses are required to establish the role of these unknown mechanisms in tumor progression.
In conclusion, the present series, the largest series in the literature, reflects the vast heterogeneity of ATCs with co-activation of multiple pathways and variable oncogenic drivers. This can partially explain the poor prognosis and the inefficiency of conventional and targeted therapies, even if other mechanisms likely contribute to the aggressive nature of ATC. They may include many abnormalities in gene expression, alterations in miRs, and epigenetic modifications that interact with mutations in oncogenes and tumor suppressors to result in such a virulent tumor. NGS analysis, by providing a better understanding of ATC oncogenesis, can inform clinical trials using a combination of targeted therapies for the development of new treatments for ATCs.
Footnotes
Acknowledgments
We warmly thank Dr. Terdjman from a private pathology lab in Valence (France) and a private pathology lab in Pau (France) for the ATC cases sent to us for this study. We would also like to thank V. Velasco for TMA conception and immunohistochemistry technics, A. Ribeiro for FISH technics, and M. Boucheix for NGS technics. Finally, the authors would like to thank Dr. Ravi Nookala of Institut Bergonié for the medical writing service.
Author Disclosure Statement
No competing financial interests exist.