
Abstract
Background:
Anaplastic thyroid carcinoma (ATC) accounts for only 3% of thyroid cancers, yet strikingly, it accounts for almost 40% of thyroid cancer deaths. Currently, no effective therapies exist. In an effort to identify ATC-specific therapeutic targets, we analyzed global gene expression data from multiple studies to identify ATC-specific dysregulated genes.
Methods:
The National Center for Biotechnology Information Gene Expression Omnibus database was searched for high-throughput gene expression microarray studies from human ATC tissue along with normal thyroid and/or papillary thyroid cancer (PTC) tissue. Gene expression levels in ATC were compared with normal thyroid or PTC using seven separate comparisons, and an ATC-specific gene set common in all seven comparisons was identified. We investigated these genes for their biological functions and pathways.
Results:
There were three studies meeting inclusion criteria, (including 32 ATC patients, 69 PTC, and 75 normal). There were 259 upregulated genes and 286 downregulated genes in ATC with at least two-fold change in all seven comparisons. Using a five-fold filter, 36 genes were upregulated in ATC, while 40 genes were downregulated. Of the 10 top globally upregulated genes in ATC, 4/10 (MMP1, ANLN, CEP55, and TFPI2) are known to play a role in ATC progression; however, 6/10 genes (TMEM158, CXCL5, E2F7, DLGAP5, MME, and ASPM) had not been specifically implicated in ATC. Similarly, 3/10 (SFTA3, LMO3, and C2orf40) of the most globally downregulated genes were novel in this context, while 7/10 genes (SLC26A7, TG, TSHR, DUOX2, CDH1, PDE8B, and FOXE1) have been previously identified in ATC. We experimentally validated a significant correlation for seven transcription factors (KLF16, SP3, ETV6, FOXC1, SP1, EGFR1, and MAFK) with the ATC-specific genes using microarray analysis of ATC cell lines. Ontology clustering of globally altered genes revealed that “mitotic cell cycle” is highly enriched in the globally upregulated gene set (44% of top upregulated genes, p-value <10−30).
Conclusions:
By focusing on globally altered genes, we have identified a set of consistently altered biological processes and pathways in ATC. Our data are consistent with an important role for M-phase cell cycle genes in ATC, and may provide direction for future studies to identify novel therapeutic targets for this disease.
Introduction
U
Large-cohort gene expression studies comparing cancer to control are powerful tools in uncovering novel oncogene pathways; however, they often suffer from lack of reproducibility. Indeed, differential genes expression in any one experiment may be more due to confounding factors than specific cancer effects. In light of this and the stark differences in survival between DTC and ATC, we sought to address this deficiency and find genes and pathways uniquely upregulated only in ATC tumors. In this study, we used gene expression data from three major studies (GSE29265, GSE33630, and GSE65144) comprising 32 ATC, 69 PTC, and 75 normal thyroid patient tissue samples. We compared gene expression profiles of ATC with normal thyroid and PTC using seven separate comparisons, and identified an ATC-specific set of genes altered in all seven comparisons. Differentially expressed genes were further annotated to biological processes, cellular components and biological pathways to identify functional aspects of dysregulated genes. These analyses revealed that cell cycle–related processes, specifically mitotic cell cycle genes, were highly enriched upregulated in ATC. In addition to confirming several known ATC-related genes, our approach of combined analyses of gene expression studies provides a novel set of genes involved in ATC that may be relevant for future therapeutic strategies in this disease.
Materials and Methods
Gene expression data analyses
The National Center for Biotechnology Information Gene Expression Omnibus (GEO) database was searched for high-throughput gene expression microarray studies that included human ATC tissue as well as normal thyroid and/or PTC tissue. Microarray studies based on immortalized ATC cell lines were specifically excluded. The three studies using an identical platform (Affymetrix Human Genome U133 Plus 2.0 Array) were selected for further analyses (GSE29265: 9 ATC, 20 PTC, 20 normal; GSE33630: 11 ATC, 49 PTC, 45 normal; and GSE65144: 12 ATC, 10 normal). Microarray raw expression data (CEL files) were downloaded from the GEO database and each dataset was analyzed separately (Fig. 1). Normalization was performed using the Robust Multiarray Average package, followed by annotation of probes to human genes using the hgu133plus2.db package. We used the Linear Models for Microarray Bioconductor package for differential expression analysis. To discover ATC-specific genes, gene expression levels in the ATC group were compared with PTC and normal groups in each dataset separately, resulting in seven different comparisons. We used the False Discovery Rate method to adjust the p-values for multiple testing, which is Benjamini and Hochberg's method to control the false discovery rate. Gene lists significantly differentially expressed in each comparison (using fold change (FC) >2.0 and adjusted p-value <0.01) were filtered. Reproducible ATC-specific genes differentially expressed in all seven comparisons (259 upregulated and 286 downregulated) were selected for further analyses (Fig. 1). In order to understand the expression patterns of differentially expressed genes, we performed cluster analysis using HPCluster program (8).
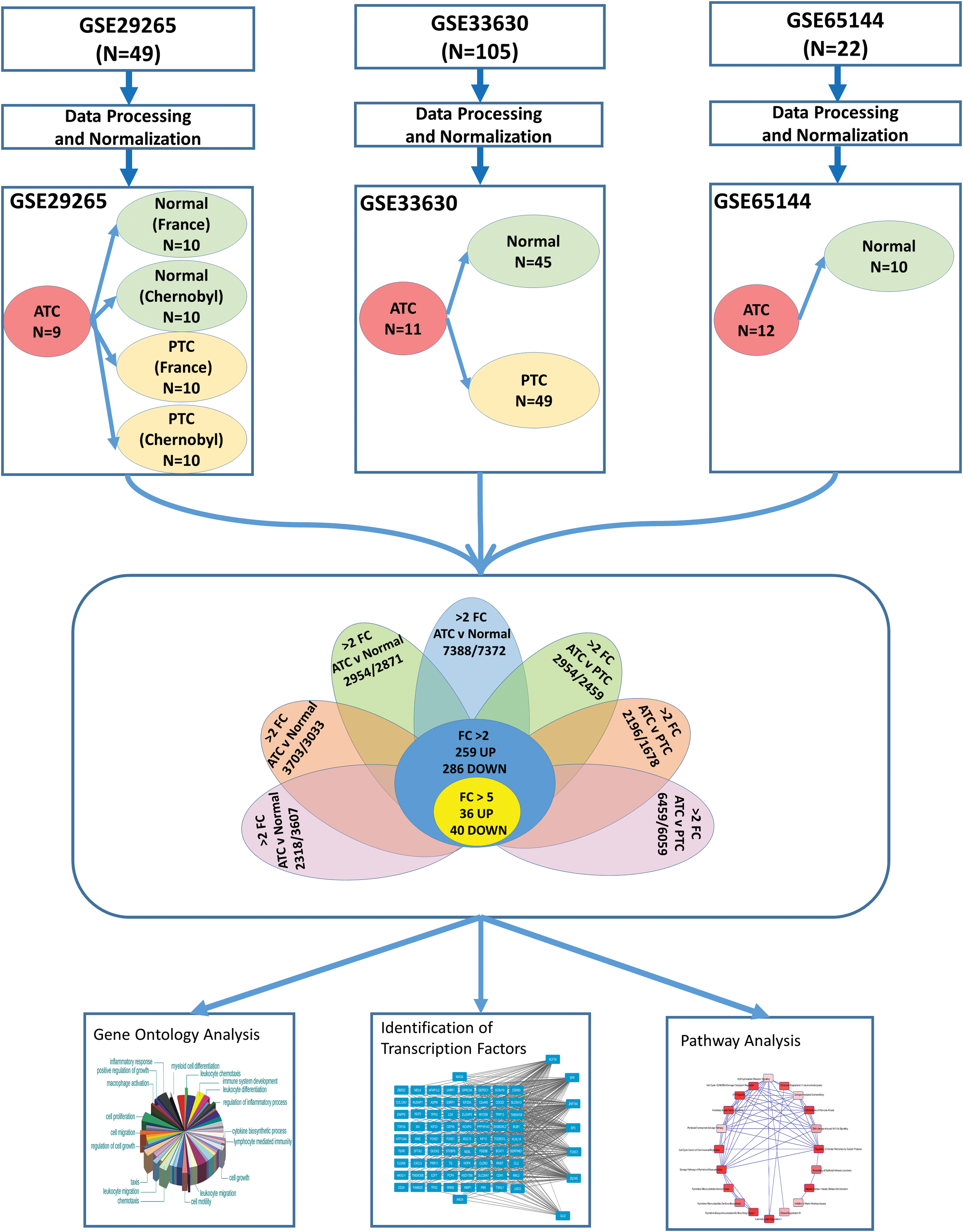
Flowchart summarizing the study design. Three studies that included human anaplastic thyroid carcinoma (ATC) tissue for gene expression microarrays were selected. Microarray raw expression data (CEL files) were downloaded from the Gene Expression Omnibus database and each dataset was analyzed separately. Seven different comparisons were made to compare ATC group with normal and papillary thyroid cancer (PTC) groups. ATC-specific genes common in all seven comparisons were identified. These genes were further investigated for their biological functions, pathways, and transcription factors. FC, fold change. Color images available online at
Biological function and pathway analyses
Gene ontology enrichment analysis was performed on 545 ATC-specific genes using DAVID (9) to provide an overview of the major biological process and cellular components. Ingenuity pathway analysis (IPA) was used to identify canonical pathways represented by the ATC-specific gene set (10). Network analysis was performed to search for defined molecular interactions between genes (gene products). To identify important transcription factors involved in the ATC-specific gene set, we used MEME and TOMTOM software tools. MEME was used to explore 5 kb upstream of specific genes for the motifs of length (6–20 bp), with stringent threshold of e-value ≤1 × 10−10. Next the TOMTOM and Jolma 2013 database was used to identify transcription factors which are likely to bind significantly (p-value ≤0.01) to their corresponding motifs (11). We used the motif–gene relationship from MEME to look into the relationship between transcription factors (TF) and gene network. For each gene, we identified upstream motifs and probable transcription factors at a p-value cut-off of 0.01. We used a custom Perl script to design a TF-gene network list and Cytoscape 3.2.1 was used to visualize this network.
Expression profiling of ATC and normal thyroid cell lines
Gene expression microarray analysis was performed for four ATC cell lines (THJ11T, THJ16T, THJ21T, THJ29T; see reference Marlow et al. (12) for characterization of these cell lines) and 3 normal primary thyroid cells (THJ-45N, THJ-101N, THJ-122N) using Affymetrix Human Genome U133 Plus 2.0 Array. Briefly, RNA was extracted from cell lines using TRIzol (Invitrogen) and chloroform (Sigma). The 18S/28S bands were verified on a 1% agarose gel. RNA quality was assessed by Agilent Bioanalyzer. The RNA products were column-purified (Affymetrix) and then in vitro transcribed to generate biotin-labeled cRNA. The in vitro transcription products were column purified, fragmented, and hybridized onto Affymetrix U133 Plus 2.0 GeneChips® at 45°C for 16 hours. Subsequent to hybridization, the arrays were washed and stained with streptavidin-phycoerythrin, then scanned in an Affymetrix GeneChip® Scanner 3000. All control parameters were confirmed to be within normal range before normalization and data reduction was initiated. Raw data was processed by MAS.5 (Affymetrix) and analyzed using GeneSpring GX10.
Results
Discovery of novel ATC-specific genes
A total of seven comparisons were made to discover genes differentially expressed in ATC as compared with normal or PTC groups (Fig. 1). Enumeration of genes with significant (adjusted p < 0.01) changes in each comparison are presented in Table 1. There were 545 genes (259 upregulated and 286 downregulated) in ATC with at least two-fold change globally present in 7/7 (100%) comparisons (Table 1). Using a more stringent five-fold filter, 36 genes were upregulated in ATC (Table 2), while 40 genes were downregulated (Table 3). The heat map representing the expression levels of 76 genes in individual samples is shown in Fig. 2. Unsupervised cluster analysis of these genes revealed three clustering patterns (Fig. 2). Cluster 1 represents a set of genes that have low expression in the majority of ATC samples and high expression in PTC and normal samples. Cluster 2 represents genes with high expression in ATC and low expression in PTC and normal samples. Cluster 3 represents genes with low expression in ATC, high expression in normal, and mixed expression in PTC samples. The 10 most upregulated genes in ATC were MMP1 (mean FC: 32.6), TMEM158 (mean FC: 23.1); CXCL5 (mean FC: 21.3); ANLN (mean FC: 17.4); E2F7 (mean FC: 14.51); DLGAP5 (mean FC: 13.0); CEP55 (mean FC: 12.57); RRM2 (mean FC: 12.5); TFPI2 (mean FC: 12.4); and MME (mean FC: 12.2). Of these top globally upregulated genes, four (MMP1, ANLN, CEP55, and COL5A1) have been previously studied in the context of ATC (13 –16); however, the remaining six genes have not been specifically implicated in ATC. The ten most downregulated genes in ATC were SLC26A7 (mean FC: −46.1); C2orf40 (mean FC: −44.4); TG (mean FC: −35.9); TSHR (mean FC: −29.9); SFTA3 (mean FC: −28.2); DUOX2 (mean FC: −27.6); CDH1 (mean FC: −25.3); PDE8B (mean FC: −22.2); LMO3 (mean FC: −21.7); and FOXE1 (mean FC: −20.5). For downregulated genes, only three (SFTA3, LMO3, and C2orf40 encoding the ECRG4 protein) were novel in this context, while the seven other genes have been explicitly proposed or studied in the context of ATC tumor biology and reviewed earlier (17).
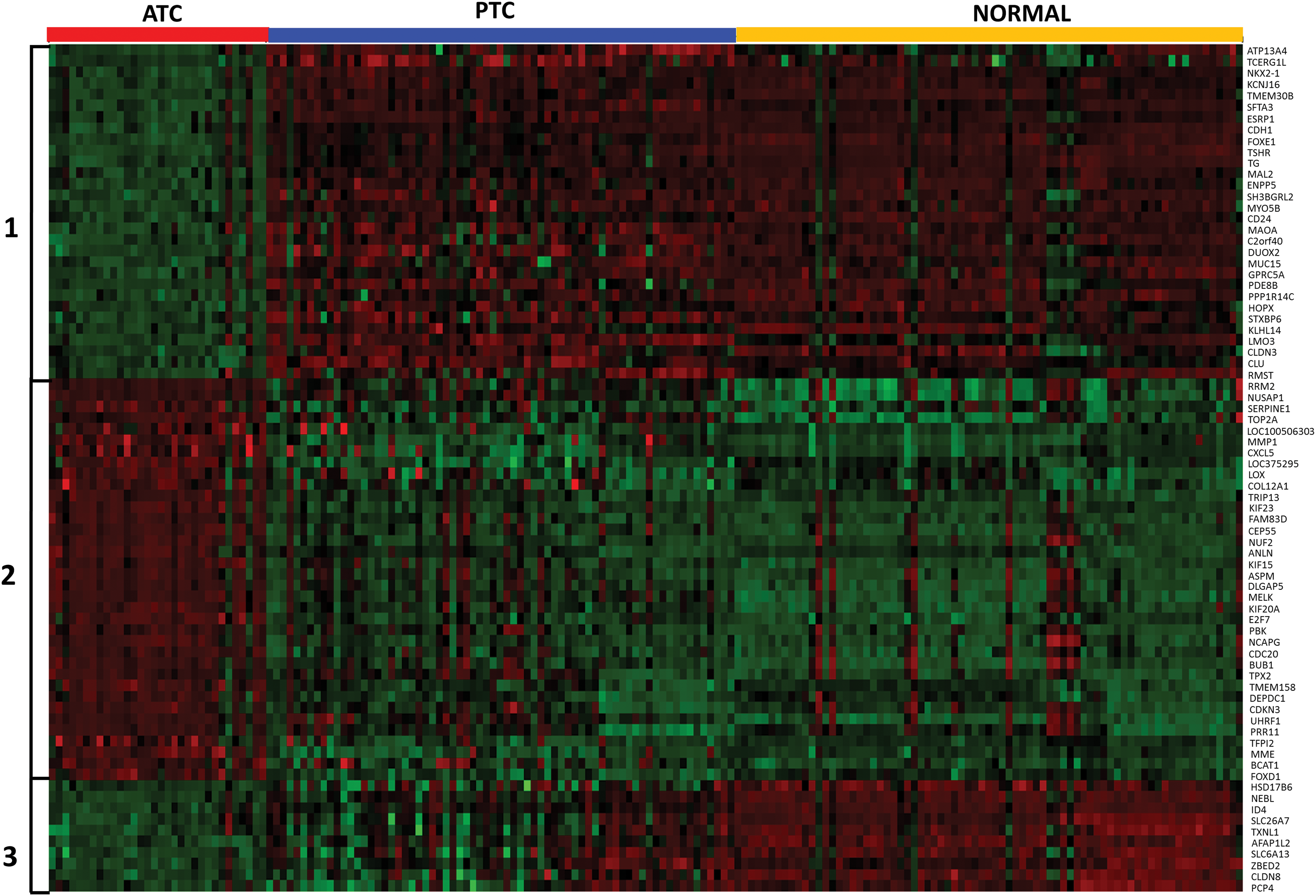
Heatmap represents clustering of 76 ATC-specific genes into three distinct clusters. Each sample is represented as a column, whereas each gene is represented as a row. Direction and magnitude of expression is represented by the color ranging from high expression (red) to low expression (green). Color images available online at
All listed genes had greater than five-fold change in all seven comparisons; False Discovery Rate (FDR) adjusted p-value <0.01.
FC1: GSE65144: ATC (n = 12) vs. Normal (n = 10).
FC2: GSE29265: ATC (n = 9) vs. PTC from Chernobyl Tissue Bank (n = 10).
FC3: GSE29265: ATC (n = 9) vs. Normal from Chernobyl Tissue Bank (n = 10).
FC4: GSE29265: ATC (n = 9) vs. PTC from the Ambroise Paré Hospital, France (n = 10).
FC5: GSE29265: ATC (n = 9) vs. Normal from the Ambroise Paré Hospital, France (n = 10).
FC6: GSE33630: ATC (n = 11) vs. Normal from Chernobyl Tissue Bank (n = 45).
FC7: GSE33630: ATC (n = 11) vs. PTC from Chernobyl Tissue Bank (n = 49).
Chr, chromosome; FC, fold change.
All listed genes had greater than five-fold change in all seven comparisons; FDR adjusted p-value <0.01.
FC1: GSE65144: ATC (n = 12) vs. Normal (n = 10).
FC2: GSE29265: ATC (n = 9) vs. PTC from Chernobyl Tissue Bank (n = 10).
FC3: GSE29265: ATC (n = 9) vs. Normal from Chernobyl Tissue Bank (n = 10).
FC4: GSE29265: ATC (n = 9) vs. PTC from the Ambroise Paré Hospital, France (n = 10).
FC5: GSE29265: ATC (n = 9) vs. Normal from the Ambroise Paré Hospital, France (n = 10).
FC6: GSE33630: ATC (n = 11) vs. Normal from Chernobyl Tissue Bank (n = 45).
FC7: GSE33630: ATC (n = 11) vs. PTC from Chernobyl Tissue Bank (n = 49).
Mitotic cell cycle is highly enriched in ATC-specific genes
Gene ontology clustering analyses was conducted to associate ATC-specific genes to different biological processes. A total of 35 biological processes were significantly enriched in globally upregulated ATC-specific genes (Table 4). Highly enriched biological processes in the globally upregulated genes are “M-phase” (50 genes, p = 2 × 10−32); “cell cycle phase” (54 genes, p = 6 × 10−32); “mitotic cell cycle” (51 genes, p = 4 × 10−31); “mitosis” (41 genes, p = 2 × 10−29); and “nuclear division” (41 genes, p = 2 × 10−29). These results are consistent with an important role for M-phase cell cycle genes in ATC. In globally downregulated genes a total of 27 biological processes were significantly enriched (Supplementary Table S1; Supplementary Data are available online at
Further analysis was conducted to associate ATC-specific genes to different cellular components. A total of 18 cellular components were significantly enriched (p < 0.05) in globally upregulated genes in ATC (Table 5). Examination of cellular components revealed that “spindle” (27 genes; p = 8 × 10−18); “microtubule cytoskeleton” (43 genes, p = 8 × 10−16); and “chromosome centromeric region” (21 genes, p = 2 × 10−12) are the most enriched cellular components in the upregulated genes, and interestingly, these cellular components are all involved in the M-phase of cell division. Taken together, these data suggest that cell cycle and specifically M-phase are biological processes critical to ATC. Analysis of downregulated genes revealed a total of 13 cellular components significantly enriched (Supplementary Table S2). The most enriched downregulated cellular components in ATC are “anchoring junction” (12 genes, p = 4.1 × 10−5); “endoplasmic reticulum” (29 genes, p = 2.8 × 10−4); and “adherens junction” (10 genes, p = 4.2 × 10−4).
Next, IPA was used to associate the 545 ATC-specific genes (including up- and downregulated) to known canonical pathways. IPA analyses revealed that the 31 pathways most affected by ATC-specific genes include canonical pathways likely to play an important role in cell cycle M-phase: “cell cycle: G2/M DNA damage checkpoint regulation” (49 genes, p = 2.0 × 10−4); “mitotic roles of polo-like kinase” (66 genes, p = 2.3 × 10−4); “salvage pathways of pyrimidine ribonucleotides” (93 genes, p = 5.4 × 10−4); “pyrimidine deoxyribonucleotides de novo biosynthesis 1” (22 genes, p = 2.0 × 10−3); “regulation of cellular mechanics by calpain protease” (57 genes, p = 3.0 × 10−3); “ATM signaling” (59 genes, p = 3.6 × 10−3); and “cell cycle control of chromosomal replication” (27 genes, p = 4.4 × 10−3) (Fig. 3). The majority of genes in these pathways showed upregulation. Together, these data reinforce the likelihood that cell cycle M-phase may play a major role in ATC tumor biology.
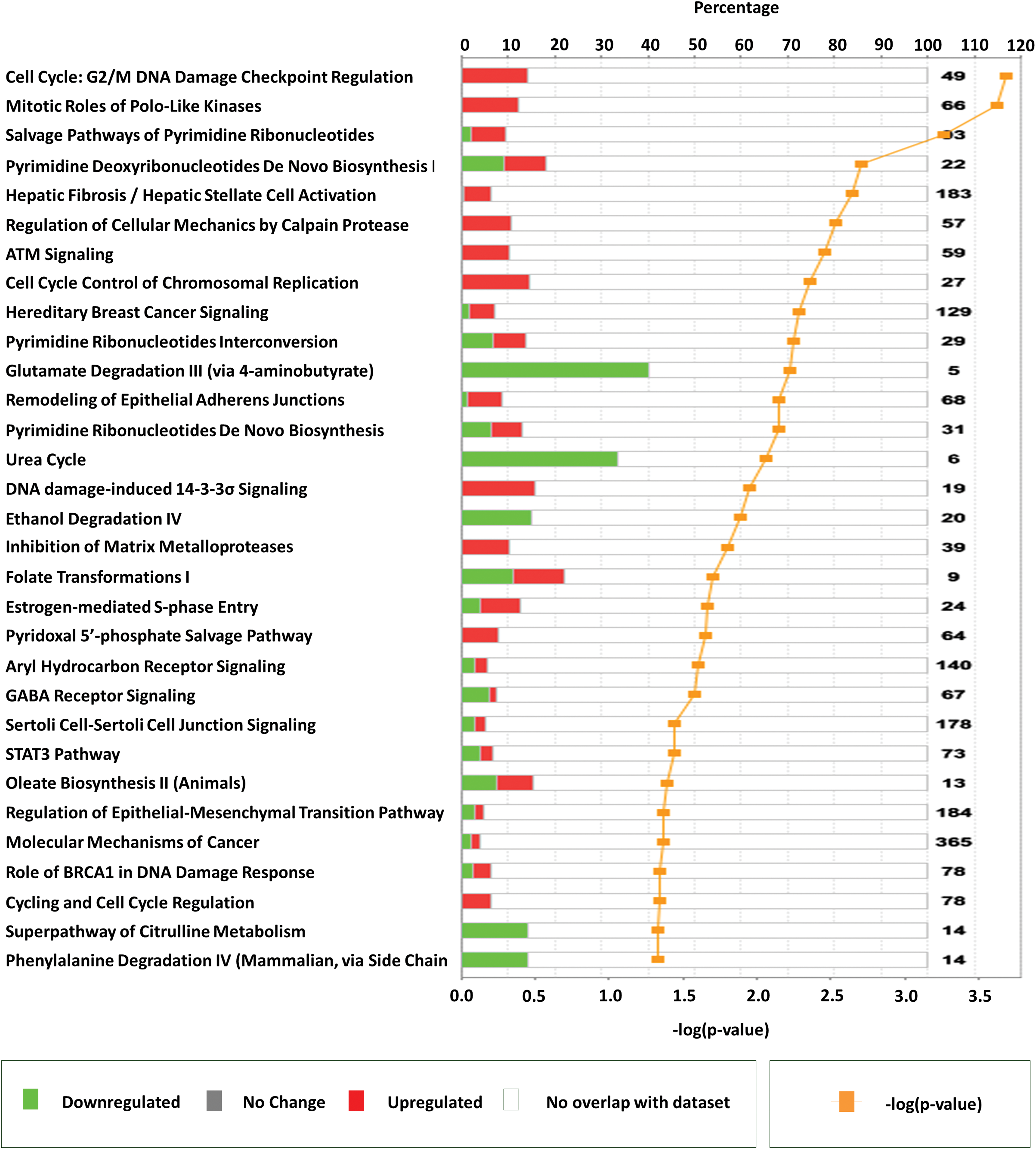
Important pathways represented by anaplastic thyroid carcinoma–specific genes identified using Ingenuity Pathway Analysis. Horizontal bars represent percentage of overlapping genes in a pathway. Red color signifies upregulation, green color signifies downregulation, and white color represents no overlap with dataset. Yellow line plots the −log (p-value) of pathway membership of genes in a specific pathway. Color images available online at
Network analysis of ATC-specific genes
IPA software was further used to explore functional relationships between these up- and downregulated genes based on known interactions using multiple datasets, resulting in 25 networks. The four highest scoring networks (score >39) are shown in Fig. 4. The top diseases and functions associated with these three networks are cancer, cell death and survival, organismal injury and abnormalities, cell cycle, cellular movement, reproductive system development and function, and cellular assembly and organization. The data on all 25 networks is presented in Supplementary Table S3.
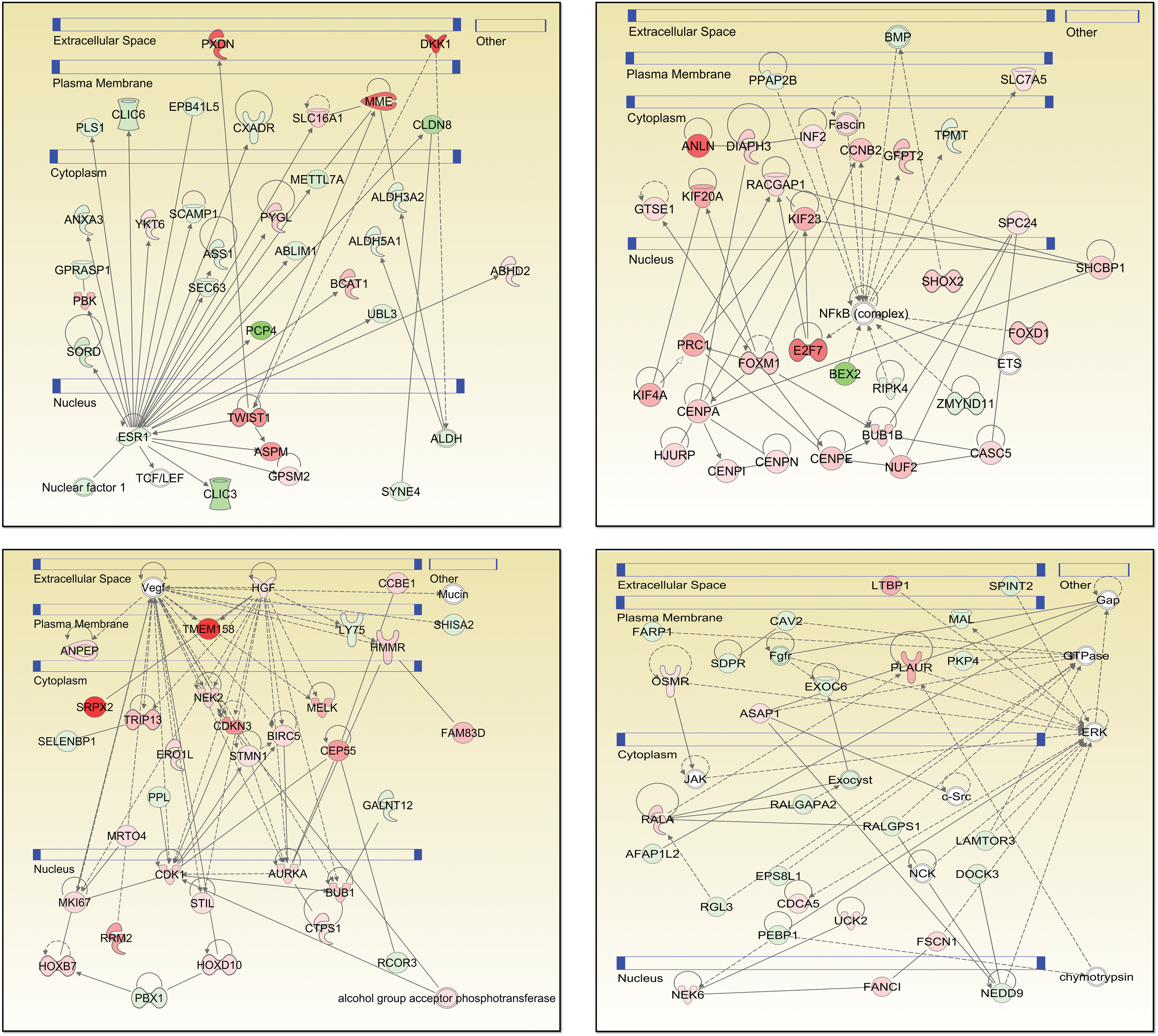
Four top-scoring networks from Ingenuity Pathway Analysis of anaplastic thyroid carcinoma specific genes. Each gene is represented as a node, and an edge represents an interaction between two nodes. Red nodes indicate upregulated genes, whereas green indicates downregulated genes. White nodes indicate genes not present in ATC-specific gene set. A solid line represents direct functional interaction, while a dotted line represents an indirect interaction. An arrow indicates action of a gene product on a target. Blue bars denote cellular compartments. Color images available online at
Identification of transcription factors
Transcription factor regulation is a critical dimension of gene expression regulation. We used MEME and TOMTOM software tools to identify transcription factors predicted to affect the expression of the 545 ATC-specific genes (see Methods). The resulting transcription-gene network was visualized using Cytoscape. There were 23 transcription factors predicted to be involved in the regulation of the 545 ATC-specific genes (Fig. 5, p-value <0.005). These 23 transcription factors were enriched for a cluster of transcription factors belonging to the SP/KLF family (SP1, SP3, SP8, KLF14, and KLF16). Other transcription factors identified in this analysis were CPEB1, EGR1, ELK1, ETV6, GLI2, FOXC1, FOXG1, MAFK, MEF2D, ONECUT1, ONECUT3, PAX1, PAX9, ZBTB49, ZIC3, ZFP740, ZNF524, and ZNF740.
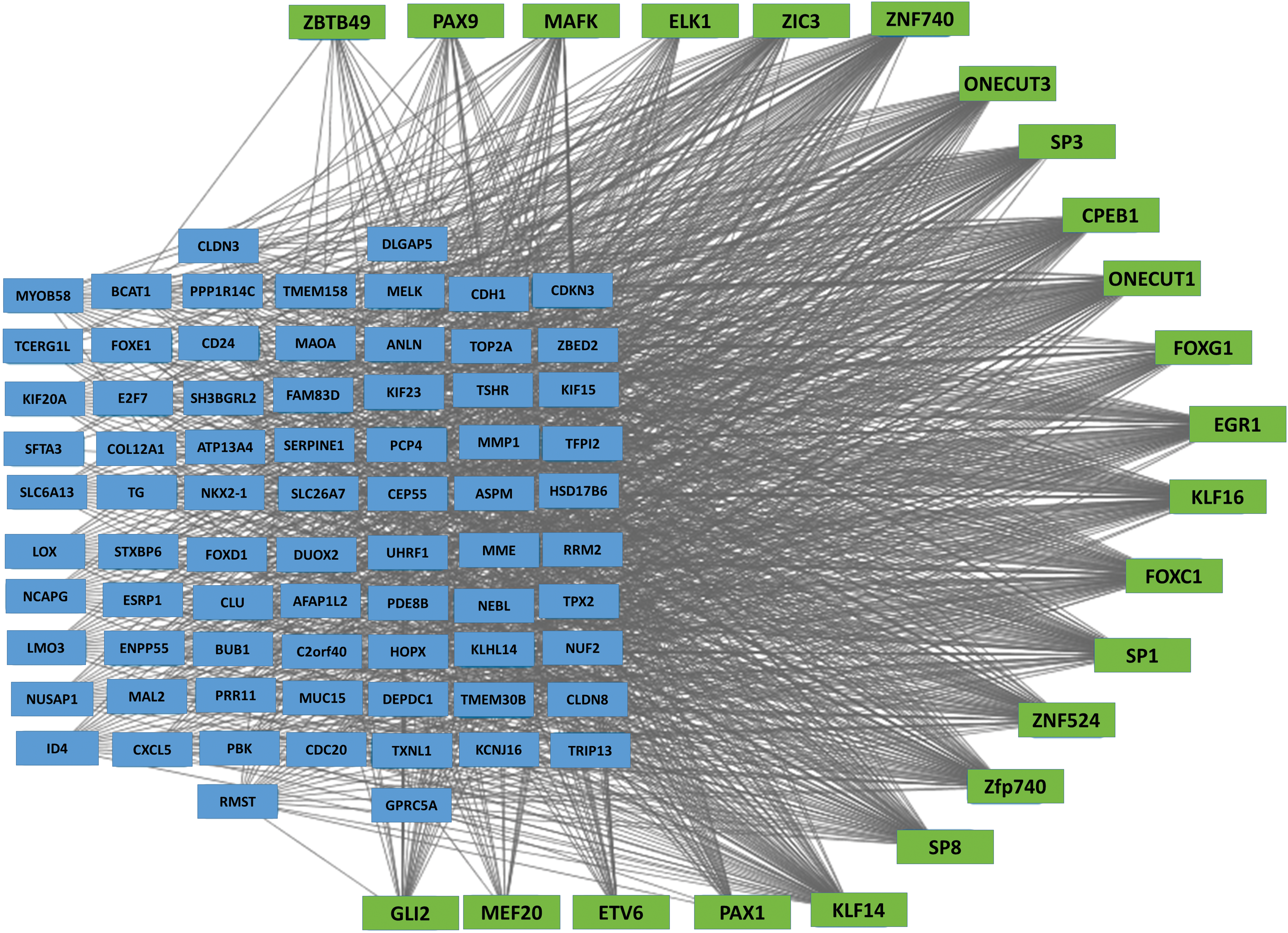
Transcription factor regulation network of ATC specific genes. Genes (blue rectangles) with at least two-fold change in expression were included in MEME and TOMTOM analysis to identify transcriptional factors (green rectangle) that regulate ATC specific genes. Color images available online at
Experimental validation of identified transcription factors
For experimental validation of identified transcription factors, we performed gene expression microarray analysis of four ATC cell lines (THJ11, THJ16, THJ21, THJ29) along with three normal primary cells (THJ45N, THJ101N, THJ122N). Correlations between expression levels of these transcription factors with their regulated ATC-specific genes were checked. We were able to validate a significant correlation for seven transcription factors (KLF16, SP3, ETV6, FOXC1, SP1, EGFR1, and MAFK) with the ATC-specific genes (Fig. 6). The cell line gene expression microarray data is submitted to the GEO database (GSE85457).
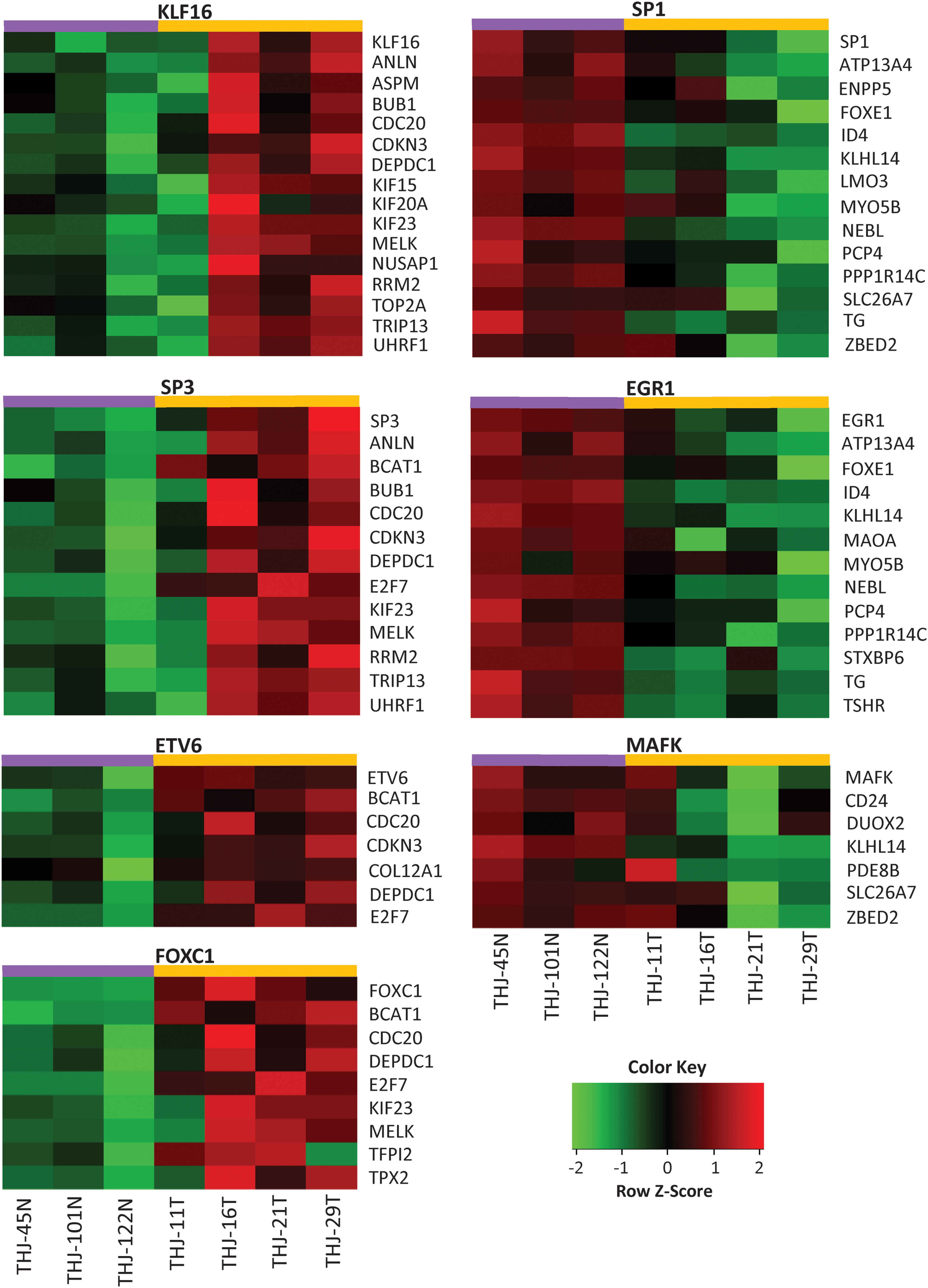
Experimental validation of identified transcription factors. Gene expression microarray analysis was performed for four ATC cell lines (THJ11, 16, 21, and 29 T cells) and 3 normal primary Thyroid cell lines (THJ45, 101, and 122 N) using Affymetrix Human Genome U133 Plus 2.0 Array. Each row represents one gene, and each column represents one cell line. Red indicates higher expression and green indicates lower expression. Color images available online at
Changes in expression of genes associated with immune infiltration
Recent studies show that immune reaction, involving tumor-associated macrophages and lymphocytic infiltration, is associated with decreased survival in advanced thyroid cancer (18,19). Furthermore, ATC tumors as a group tend to have significant macrophage infiltration, up to 50% of tumor mass in some cases (20). To place our data in context with these findings, we compiled a list of genes associated with “tumor associated macrophages,” “lymphocytic infiltration,” and “phagocytosis by macrophages” (Table 6). We then analyzed the expression of these genes in our ATC samples. We found 1 gene significantly downregulated (FC <0.5-fold) and 27 genes significantly upregulated (FC >2-fold) in ATC samples (Fig. 7). CSF1R (mean FC: 3.6) encodes for the cell surface receptor for colony stimulating factor 1 (CSF1), a cytokine which controls the production, differentiation, and function of macrophages. Other immune related genes highly upregulated in ATC are CXCL5 (mean FC: 21.3); SERPINE1 (mean FC: 10.1); THBS1 (mean FC: 6.9); and GJB2 (mean FC: 5.7); LGALS1 (mean FC: 5.4); CCR1 (mean FC: 5.2); HGF (mean FC: 3.7); and CLIC4 (mean FC: 3.0). Several members of a family of immunoglobulin Fc receptor genes including FCGR2A (mean FC: 5.8); FCER1G (mean FC: 4.8); and FCGR2B (mean FC: 3.1) were also upregulated in ATC (Table 6).
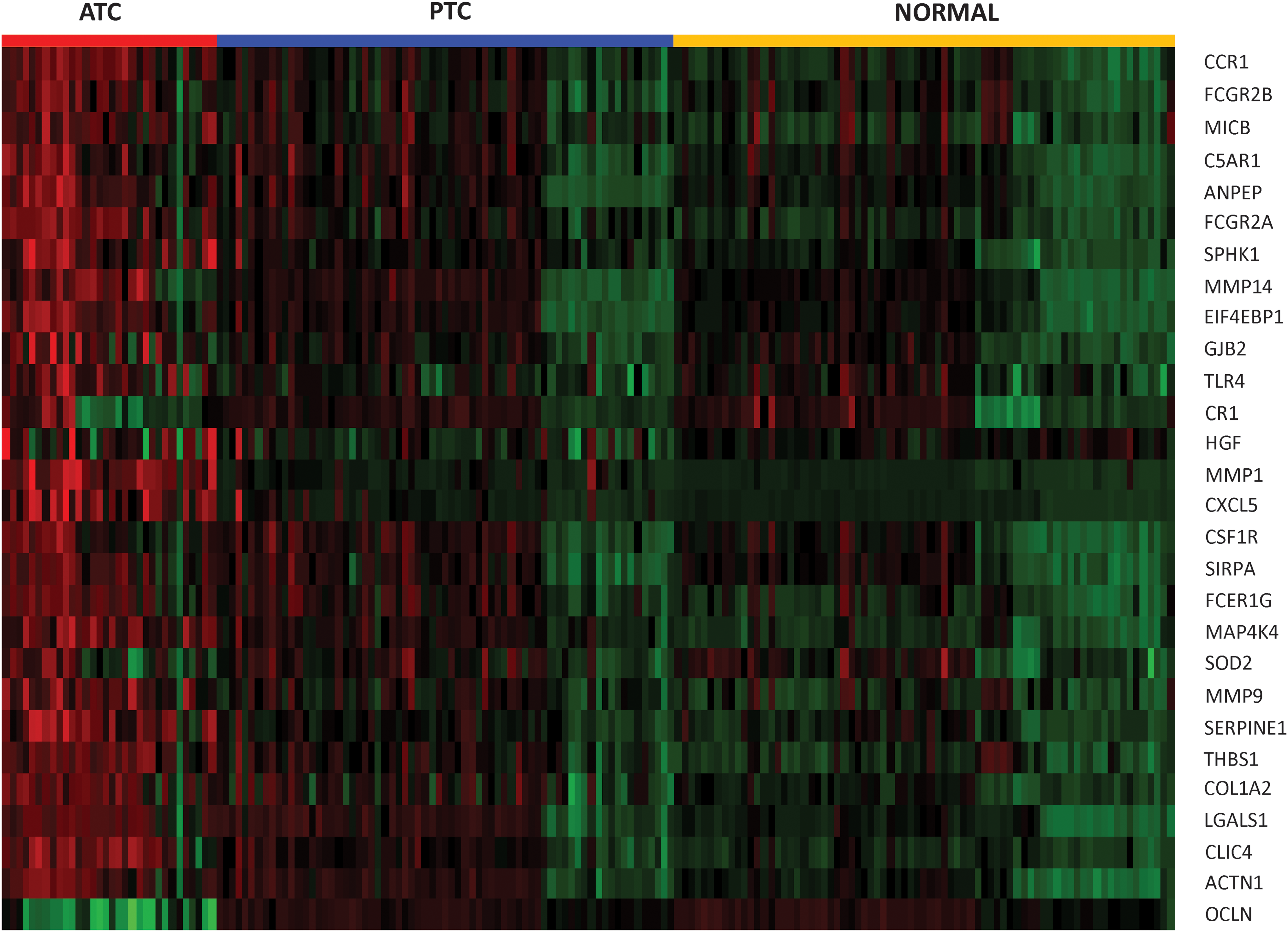
Heatmap representing 28 immune related genes significantly changed in ATC. Each sample is represented as a column whereas each gene is represented as a row. Direction and magnitude of expression is represented by the color ranging from high expression (red) to low expression (green). Color images available online at
All listed genes had greater than two-fold change in all seven comparisons; FDR adjusted p-value <0.05.
FC1: GSE65144: ATC (n = 12) vs. Normal (n = 10).
FC2: GSE29265: ATC (n = 9) vs. PTC from Chernobyl Tissue Bank (n = 10).
FC3: GSE29265: ATC (n = 9) vs. Normal from Chernobyl Tissue Bank (n = 10).
FC4: GSE29265: ATC (n = 9) vs. PTC from the Ambroise Paré Hospital, France (n = 10).
FC5: GSE29265: ATC (n = 9) vs. Normal from the Ambroise Paré Hospital, France (n = 10).
FC6: GSE33630: ATC (n = 11) vs. Normal from Chernobyl Tissue Bank (n = 45).
FC7: GSE33630: ATC (n = 11) vs. PTC from Chernobyl Tissue Bank (n = 49).
Ig, immunoglobin; MHC, major histocompatibility complex.
Discussion
Our study was designed to provide new insights into ATC-specific tumor biology in humans. By leveraging publicly available gene expression data to identify genes altered uniquely and reproducibly in ATC, we provide evidence that cell cycle control genes are dysregulated in ATC. Several previous ATC studies have attempted to leverage global gene expression assays such as expression microarrays to investigate gene dysregulation in ATC. Three of these provided the raw data for the present study. In 2012, Hebrant et al. (data deposited online as GSE33630) compared mRNA expression profiles of 11 ATC, 49 PTC tumors, and 45 adjacent normal thyroids and found that the majority of genes altered in ATC were also altered in PTC (16). They identified a nine-gene signature that clustered ATC from PTC, consisting of downregulation of NELL2, SPINT2, MARVELED2, DUOXA1, RPH3AL, TBX3, PCYOX1, C5orf41, and PKP4 in ATC. These were subsequently confirmed using RT-PCR. Tomas et al. compared mRNA expression in 9 ATCs, 20 PTCs, and 20 normal thyroids in an effort to identify Chernobyl-specific gene expression signatures (data deposited online as GSE29265), but there is no corresponding manuscript at this time. Von Roemeling et al. (data deposited online as GSE65144) examined mRNA expression signatures from 12 ATC and 13 normal thyroids (21). They performed extensive validation directed toward alterations in fatty acid metabolism identified by the gene expression study but did not perform gene ontology or pathway clustering. There are also additional genomic or transcriptomic ATC studies leveraging different technologies that have been reported in the literature (22 –29). These were not included, as our methods necessitated using data derived from a single platform. It is, however, notable that to date none of these individual studies have led to a therapy for ATC that has demonstrated improved survival in clinical trials.
Therefore, in an effort to identify more robust ATC-specific therapeutic targets, we analyzed data from multiple gene expression microarray studies to identify globally altered genes. Combining microarray gene expression data from multiple laboratories or array platforms can have confounding batch effects leading to false discoveries (30). Many methods exist for removing batch effects from data, however, batch adjustments may bias the results and systematically induce incorrect group differences in downstream analyses (31). Rather than follow a more traditional approach (combined analysis), we instead performed separate comparisons in each dataset followed by identification of statistically significant, differentially expressed genes, which repeatedly appeared in each comparison. We identified a gene signature comprising 259 upregulated and 286 downregulated genes with at least two-fold change globally present in all seven comparisons. This gene set was able to differentiate ATC samples from PTC and normal samples, and could have possible utility as a molecular diagnostic tool in discerning ATC from poorly differentiated PTC, although this is merely a conjecture at this point.
Our data are consistent with an important role for control of the M-phase of the cell cycle in ATC tumor biology, in that a surprising concentration of ATC-specific genes impinge on this pathway. To our knowledge, this is the first report that reveals dysregulation of this pathway in ATC. This is particularly interesting in light of the fact that our analysis made use of publicly available datasets; these were deposited after analysis and reporting of the individual data. As such, the individual up- and downregulated genes were, by definition, altered in these original datasets. In many cases, however, they were “buried” in the middle of the data as altered but not in the “top” list either by p-value or fold-change value. Our analysis focused attention from these several thousand potentially important genes identified in each individual comparison to a manageable and enriched set of ∼500 (FC >2) and ∼70 (FC >5) genes. This supports our hypothesis that our combinatorial approach may help to focus attention toward genes that are reproducibly and consistently altered, and thus may be imputed to possibly hold an increased likelihood of playing an important functional role.
Internal validation of our approach is provided in that among the top 10 over- and under-expressed ATC-specific genes, 40% and 70% respectively, have been the focus of previous investigations in ATC. For example, thyroglobulin (TG) and thyrotropin receptor (TSH-R) are universally lost in ATC (17). CDH1, an important regulator of epithelial-mesenchymal transition, has likewise been shown to be downregulated in ATC (32,33). Conversely, matrix metalloproteinase-1 (MMP1), important for allowing invasion and metastasis, is highly overexpressed in ATC (16,34). Poorly differentiated thyroid carcinoma specimens have profound deregulation of genes involved in cell adhesion and intracellular junctions, with changes consistent with an epithelial–mesenchymal transition (35). A significant upregulation of the “cell cycle progression” was found by the functional profiling of undifferentiated and well-differentiated thyroid tumors (33). These confirmatory findings notwithstanding, the more interesting genes are the ones not yet described in the context of ATC tumor biology, as these may suggest novel therapeutic targets or strategies for this disease. A few of the more promising leads will be further explored here, although space limitations preclude an exhaustive analysis.
One highly upregulated gene, novel in the context of ATC, was C-X-C motif chemokine 5 (CXCL5). CXCL5 is known to play a role in macrophage regulation, and ATC tumors tend to have significant macrophage infiltration. As our study utilized publicly available datasets, determination of macrophage involvement in the ATC tumors included in this study is not possible. It is likely that the CXCL5 upregulation may reflect inclusion of macrophage RNA in the analyses, but it is also possible that CXCL5 also may play a role in ATC tumor biology. CXCL5 has recently been implicated in mediating cell proliferation, migration and invasion in colorectal cancer and is associated with metastasis and worse prognosis in gastric and breast cancer (36,37). Another novel, ATC-specific overexpressed gene encodes the disks large-associated protein 5 (DLGAP5), a component of the kinetochore responsible for stabilizing microtubules and the spindle apparatus. This protein is relatively unstudied in tumor biology, but recently was shown to have a potential role in hepatocellular cancer. Liao et al. demonstrated overexpression of DLGAP5 mRNA to be common in tumor samples from liver cancer patients, and that in vitro RNA-interference mediated silencing of DLGAP5 inhibited proliferation and invasion (38). One of the novel, ATC-specific underexpressed genes was C2orf40, which encodes the esophageal cancer–related gene 4 (ECRG4) protein, a tumor suppressor affecting cancer cell migration, invasion and cell cycle regulation and downregulated in esophageal and breast cancer (39,40).
Our transcription factor analysis identified several transcription factors likely to be important in ATC tumor biology. We performed further experimental validation of the identified transcription factors using four ATC cell lines along with three normal primary cells. We were able to validate a significant correlation for seven transcription factors (KLF16, SP3, ETV6, FOXC1, SP1, EGFR1, and MAFK) with the ATC-specific genes (Fig. 6). Specificity-Protein/Krueppel Like Factor (SP/KLF) transcription factors (including SP1, SP3, and KLF16 identified in the present study) comprise an emerging group of proteins that are thought to regulate fundamental cellular processes including cell cycle and growth control, metabolic pathways, and apoptosis. There is emerging evidence that expression of genes known to play pivotal roles in metastasis and cell proliferation are regulated by the Sp family of proteins (41). NTRK3/ETV6 is a known fusion oncogene (42,43) and results in more extensive disease and aggressive pathology of PTC in the pediatric population (44). FOXC1 is a member of the Forkhead box family transcription factors and is known to promote melanoma by activating the MST1R/PI3K/AKT pathway (45). Although transcription factors have traditionally been considered undruggable targets, recent successes in this arena call this into question (46 –49). Taken together, these data suggest that pharmacologic targeting of transcription factors upregulated in ATC (KLF16, SP3, ETV6, and FOXC1) might be an area for future investigations in developing novel ATC therapeutics.
Thyroid cancers are heavily infiltrated with macrophages (18) and the density of tumor-associated macrophages is increased in advanced thyroid cancers including ATC (19). The presence of a high density of macrophages may influence the overall gene expression profile in ATC. Several upregulated genes encoding cytokine, chemokine and matrix metalloproteinases are highly expressed by tissue resident macrophages. We found several genes associated with “tumor-associated macrophages (TAMs),” “lymphocytic infiltration” and “phagocytosis by macrophages” including CSF1R (mean FC: 3.6), highly upregulated in ATC samples (Table 6). Macrophages depend on CSF-1 for differentiation and survival. The CSF1R inhibitor has been used to target TAMs in a mouse proneural GBM model, which significantly increased survival and regressed established tumors (50). TAMs are emerging as a promising therapeutic target and our results suggest that CSF1R inhibition could have translational potential for ATC treatment.
In conclusion, we have demonstrated that multiple cell cycle M-phase genes are highly upregulated in ATC. Several of the genes identified as ATC-specific are novel in the context of ATC tumor biology and provide a strong rationale supporting their potential as possible therapeutic targets in ATC. Additionally, transcription factor regulation of ATC-specific gene sets suggest the SP/KLF transcription factor family as additional potential therapeutic targets. Our data strongly suggest that therapeutic strategies targeting processes critical for cell cycle mitosis may be of particular value in ATC and deserve further investigation.
Footnotes
Acknowledgments
This study was funded by institutional startup funds (A.S. and P.M.W.). This work was supported in part by National Institutes of Health; National Cancer Institute Grant R01CA136665 (to J.A.C. and R.C.S.); Florida Department of Health Bankhead-Coley Cancer Research Program [Grants FL09B202 (to J.A.C. and R.C.S.) and FL3BF01 (to J.A.C.)].
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.