
Abstract
Background:
Thyroid hormone (TH) has important roles in regulating hepatic metabolism. It was previously reported that most hepatic genes activated by a single triiodothyronine (T3) injection became desensitized after multiple injections, and that approximately 10% of target genes did not return to basal expression levels after T3 withdrawal, despite normalization of serum TH and thyrotropin (TSH) levels. To determine the possible mechanism(s) for desensitization and incomplete recovery of hepatic target gene transcription and their effects on metabolism, mRNA and/or protein expression levels of key regulators of TH action were measured, as well as metabolomic changes after chronic T3 treatment and withdrawal.
Methods:
Adult male mice were treated with daily injections of T3 (20 μg/100 g body weight) for 14 days followed by the cessation of T3 for 10 days. Livers were harvested at 6 hours, 24 hours, and 14 days after the first T3 injection, and at 10 days after withdrawal, and then analyzed by quantitative reverse transcription polymerase chain reaction, Western blotting, and metabolomics.
Results:
Although TH receptor (TRα and TRβ) mRNAs decreased slightly after chronic T3 treatment, only TRβ protein decreased before returning to basal expression level after withdrawal. The expression of other regulators of TH action was unchanged. TRβ protein expression was also decreased in adult male monocarboxylate transporter-8 (Mct8)-knockout mice, an in vivo model of chronic intrahepatic hyperthyroidism. Previously, increased hepatic long-chain acylcarnitine levels were found after acute TH treatment. However, in this study, long-chain acylcarnitine levels were unchanged after chronic T3, and paradoxically increased after T3 withdrawal. Pathway analyses of the previous microarray results showed upregulation of lipogenic genes after acute T3 treatment and withdrawal. Phosphorylation of acetyl-CoA carboxylase also decreased after T3 withdrawal.
Conclusions:
Decreased hepatic TRβ protein expression occurred after chronic T3 exposure in adult male wild-type and Mct8-knockout mice. Gene array pathway and metabolomics analyses showed abnormalities in hepatic lipogenic gene expression and acylcarnitine levels, respectively, after withdrawal, despite normalization of serum TSH and TH levels. These findings may help explain the variable clinical presentations of some patients during hyperthyroidism and recovery, since TRβ protein, target gene expression, and metabolic adaptive changes can occur in individual tissues without necessarily being reflected by circulating TH and TSH concentrations.
Introduction
I
TH has important roles in regulating hepatic fatty acid, cholesterol, and carbohydrate homeostasis (6,7). Indeed, recent metabolomics analyses showed that acute TH treatment increased acylcarnitines in the mouse liver (8). To understand further the sensitivity of hepatic target genes to TH during chronic hyperthyroidism and recovery, global expression profiles of hepatic triiodothyronine (T3)-target genes were recently determined after a single liothyronine (LT3) injection (acute T3) or daily injections for 14 days (chronic T3) followed by cessation of LT3 for 10 days (T3 withdrawal) (9). Interestingly, most genes that were positively or negatively regulated by acute T3 treatment became desensitized to T3 after chronic treatment. Also, about 10% of hepatic target genes regulated either acutely or chronically did not return to basal levels of expression 10 days after stopping injections, despite allowing serum TH and thyrotropin (TSH) levels to normalize.
In order to determine the possible mechanism of desensitization and incomplete recovery of TH-regulated target genes identified in an earlier study, mRNA and/or protein expression levels of key regulators for TH action were measured during chronic T3 and after T3 withdrawal. Metabolomics changes during these conditions were also analyzed in order to understand the physiological relevance of the differential expression of T3-target genes during these different thyroid states.
Methods
Animal experimental protocol
Subsets of animal samples described previously were used (9). In brief, C57BL/6 male mice were maintained on a 12-hour light/dark cycle and had access to food and water ad libitum. Mice between the ages of 8 and 10 weeks were treated with 20 μg of liothyronine (LT3) per 100 g of body weight for 14 days followed by cessation of LT3 for 10 days. Mice were sacrificed at each of the indicated time points (Fig. 1). Both a single and multiple LT3 injections induced significant elevations (10- to 30-fold) in serum total T3 concentrations, as well as significant reductions in serum TSH concentrations (9). Serum total T3 and TSH concentrations returned to near basal levels 10 days after T3 withdrawal (p = 0.755 and p = 0.470, respectively). Mice appeared healthy, and there were no significant changes in body weight during the experimental protocol. This animal protocol was approved by the Institutional Animal Care and Use Committee of the Biopolis Resource Centre, Agency for Science and Technology (A*STAR), Singapore.
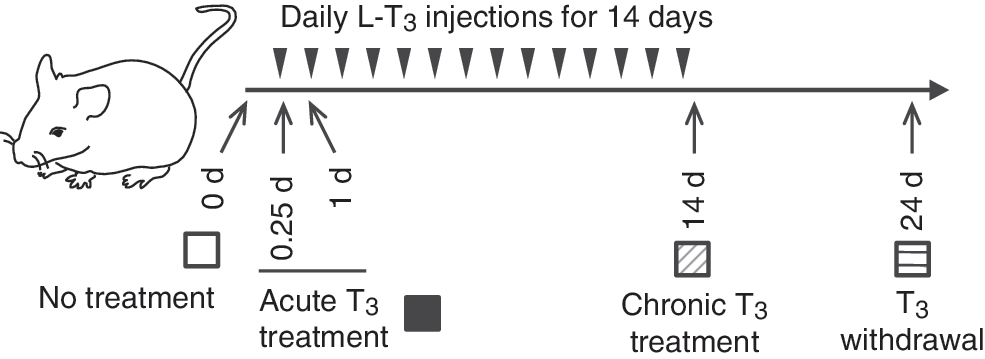
Design of animal experiments to study thyroid hormone treatment and withdrawal. Adult male mice (8–10 weeks old) were treated with 20 μg of liothyronine (LT3) per 100 g of body weight for 14 days followed by cessation of LT3 for 10 days. Mice were sacrificed at each of the indicated time points. Arrowheads represent LT3 injections. No treatment (blank); acute T3 treatment (black); chronic triiodothyronine (T3) treatment (hashed); T3 withdrawal (striped) are shown.
Monocarboxylate transporter-8 (Mct8)-knockout mice, which have chronic intrahepatic hyperthyroidism since birth, were generated, housed, and genotyped, as described previously (10). This animal protocol was approved by the University of Chicago Institutional Animal Care and Use Committee.
Cell culture and genetic knockdown using siRNA
HepG2 cells expressing TRβ (THRB-HepG2; a kind gift from Prof. Martin L. Privalsky, University of California, Davis, David, CA), which has been characterized previously (11), were cultured and maintained, as described elsewhere (8). THRB-HepG2 cells were transfected in 100 mm culture dishes, using lipofectamine RNAiMAX with 0.1–10 nM of siRNA targeting THRB (Ambion, Inc.; ID: s14119) according to the manufacturer's protocol. After 48 h of transfection, cells were subjected to T3 (10 nM) treatment in normal T3-depleted medium. After 24 h of treatment, protein lysate and total RNA were isolated for further analysis, as described previously (8).
RNA isolation and quantitative reverse transcription polymerase chain reaction
Total RNA was extracted from the liver (15–20 μg) with TRIzol reagent (Invitrogen) followed by InviTrap spin cell RNA Minikit (Stratec Molecular) according to the manufacturer's instructions. RNA concentration was measured by NanoDrop8000 (Thermo Scientific), and cDNA was reverse transcribed from 500 ng of total RNA using high-capacity cDNA reverse transcription kits (Applied Biosystems). The quantitative reverse transcription polymerase chain reactions (qRT-PCRs) were performed using the QuantiFast SYBR Green PCR Kit (QIAGEN) on the 7900HT Fast Real-Time PCR system (Applied Biosystems). Relative mRNA levels were calculated using the 2–ΔΔCt method and normalized to suitable reference genes following TH exposure (12). The stabilities of 18S ribosomal RNA (Rn18S), peptidylprolylisomerase A (Ppia), and β-actin (Actb) were evaluated under the different T3 treatment conditions, revealing Rn18S and Ppia transcripts were less responsive to T3 in liver and kidney samples, respectively (data not shown). Data are expressed as the fold change relative to the mean value of non-treated mice. The specific primer sequences used were as follows: thyroid hormone receptor beta (Thrb), forward, 5′-GAG ACT CTA ACT TTG AAT GGG-3′, reverse, 5′-CGA TCT GAA GAC ATT AGC AG-3′; thyroid hormone receptor alpha (Thra), forward, 5′-CAT GGA CTT GGT TCT AGA TG-3′, reverse, 5′-CTG TAG CAA CAT GTA TCA GG-3′; solute carrier family 16 (monocarboxylic acid transporters), member 2 (Slc16a2, or Mct8), forward, 5′-CGT GCA CCT GAT GAA ATA TG-3′, reverse, 5′-GAT CAT CAT GGA CAT CAA GC-3′; Slc16a10, or Mct10, forward, 5′-AAG CTC CAT CGA GCC TCT GTA-3′, reverse, 5′-GTC CCA AAA TGA CCA GTG ACG-3′; Dio2, forward, 5′-CAG TCT TTT TCT CCA ACT GC-3′, reverse, 5′-CCA GTT TAA CCT GTT TGT AGG-3′; Dio3, forward, 5′-AGA AAG TCA AAG GTT GTG G-3′, reverse, 5′-AAA ACG TAC AAA AGG GAG TC-3′; Rxra, forward, 5′-TAA CAG AGC TGG TGT CTA AG-3′, reverse, 5′-TTA GAG TCA GGG TTG AAC AG-3′; nuclear corepressor 1 (Ncor1), forward, 5′-AAC ATC AAG ACT GGA GTA CC-3′, reverse, 5′-ATG ATA AAA TGA TCC GTG GC-3′; and nuclear receptor coactivator 1 (Ncoa1), forward, 5′-GAC CAA GCA AGA TAC TAC AG-3′, reverse, 5′-GAG TCA TCA CTT CTT GAA ACA G-3′; solute carrier organic anion transporter family, member 1c1 (Slco1c1 or Oatp1c1), forward, 5′-GGG CCA TCC TTT ACA GTC GG-3′, reverse, 5′-CCT TCT CTC TAT CTG AGT CAC GG-3′; thyroid hormone-responsive protein (Thrsp or Spot14), forward, 5′-ACA GAC ACT GGG GAC CAA AC-3′, reverse, 5′-AGG CTT TTG AGC AGA CAG CA-3′; phosphoenolpyruvate carboxykinase 1 (Pck1), forward, 5′-AAT ATG ACA ACT GTT GGC TG-3′, reverse, 5′-AAT GCT TTC TCA AAG TCC TC-3′; Rn18S, forward, 5′-TTC CGA TAA CGA ACG AGA CTC T-3′, reverse, 5′-TGG CTG AAC GCC ACT TGT C-3′; Ppia, forward, 5′-TGT GCC AGG GTG GTG ACT TTA-3′, reverse, 5′-TGC CTT CTT TCA CCT TCC CAA A-3′; PCK1, forward, 5′-CAG CGA CGG GGG CGT TTA CT-3′, reverse, 5′-GGG CAC AAG GTT CCC CAT CCT CT-3′; POLR2A, forward, 5′-AAG ATC AAT GCT GGT TTT GG-3′, reverse, 5′-CAT CAT CAT CCA TCT TGT CC-3′.
Western blot analysis
Protein lysates from frozen liver were prepared using radioimmunoprecipitation assay (RIPA) lysis buffer, and protein concentrations were measured using the BCA kit (Bio-Rad). Immunoblotting was performed, as described previously (8). In brief, equal amounts of protein were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred immediately onto polyvinylidenedifluoride membranes (Bio-Rad). The membranes were blocked with 5% nonfat dry milk, followed by overnight incubation with the appropriate diluted primary antibodies for TRβ (sc-738; Santa Cruz), TRα (PA1-211A; Thermo Scientific), NCoR1 (A301-145A; Bethyl), NCoA1, or SRC-1 (sc-8995; Santa Cruz), RXRα (#3085; Cell Signaling), acetyl-CoA carboxylase (ACC; 3662S; Cell Signaling Technology), ACC phosphorylated at serine 79 (pACC; 3661S; Cell Signaling Technology), β-Tubulin (2128S; Cell Signaling Technology), and GAPDH (2118L; Cell Signaling Technology). Membranes were washed three times in TBST (Tris-buffered saline, 0.1% Tween 20) and subsequently incubated with species-appropriate, peroxidase-conjugated secondary antibodies (Santa Cruz) for one hour. Blots were washed three times with TBST and developed using an enhanced chemiluminescence system (GE Healthcare). Representative images of blots for TRβ and TRα are shown in Supplementary Figure S1 (Supplementary Data are available online at
Metabolomics analysis
Acylcarnitines, amino acids, and organic acids were measured in liver extracts by previously described mass spectrometry (MS)-based methods (13). Analysis employed tandem MS with a Quattro Micro instrument (Waters Corporation).
Serum triglyceride quantification
Hepatic triglyceride was quantified using the Triglyceride Colorimetric Assay Kit (Cayman), according to the manufacturer's instructions.
Statistics
All data represent the mean ± standard error of the mean. Statistical significance of differences (p < 0.05) was examined by two-tailed Student's t-test or one-way analysis of variance followed by Fisher's protected least significant difference using GraphPad PRISM v7.0 (GraphPad Software, Inc.).
Results
First, mRNA levels of several key regulators for T3-dependent transcription after chronic T3 treatment and T3 withdrawal were determined by qRT-PCR. Both acute and chronic T3 significantly reduced hepatic TRβ (Thrb) transcripts by up to 22%, whereas only chronic T3 decreased TRα (Thra) transcripts (Fig. 2). Both Thrb and Thra transcripts returned to baseline levels 10 days after T3 withdrawal. No significant changes were seen either acutely or chronically for Mct8, Mct10, Rxra, Ncor1, Ncor2 (or Smrt), or Ncoa1 transcripts (Supplementary Fig. S2A). Dio2 and Slco1c1 (or Oatp1c1) transcripts were undetectable by qRT-PCR. In earlier microarray analyses (9), no significant changes were observed in Rxrb, Rxrg, Src2, Src3, or Dio3 transcripts during either T3 treatment conditions (Supplementary Table S1).
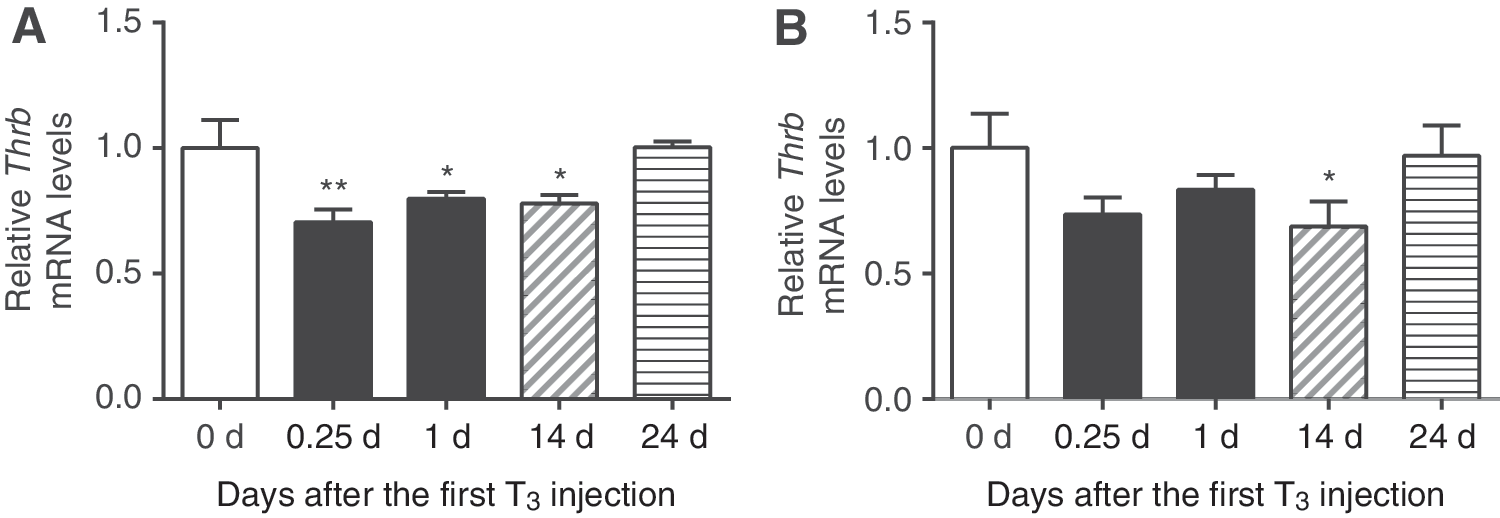
Gene expression of TR isoforms in mouse liver after T3 treatment and its withdrawal. Adult male mice (8–10 weeks old) were treated with 20 μg of LT3/100 g body weight for 14 days followed by cessation of LT3 for 10 days. Relative mRNA expression of liver Thrb (
Next, the protein expression of TR isoforms was analyzed. Interestingly, T3 decreased TRβ protein expression in a time-dependent manner (Fig. 3A), and was decreased 66% 14 days after the first T3 injection. In contrast, no changes in TRα proteins levels were detected (Fig. 3B). Moreover, no significant changes were seen in NCoR1, NCoA1, or RXRα protein expression after T3 treatment (Supplementary Fig. S2B). TRβ protein expression returned to near basal levels 10 days after the last (14th) LT3 injection (Fig. 3A) and was consistent with the return to basal levels of most acutely and chronically regulated target genes during that time period (9).
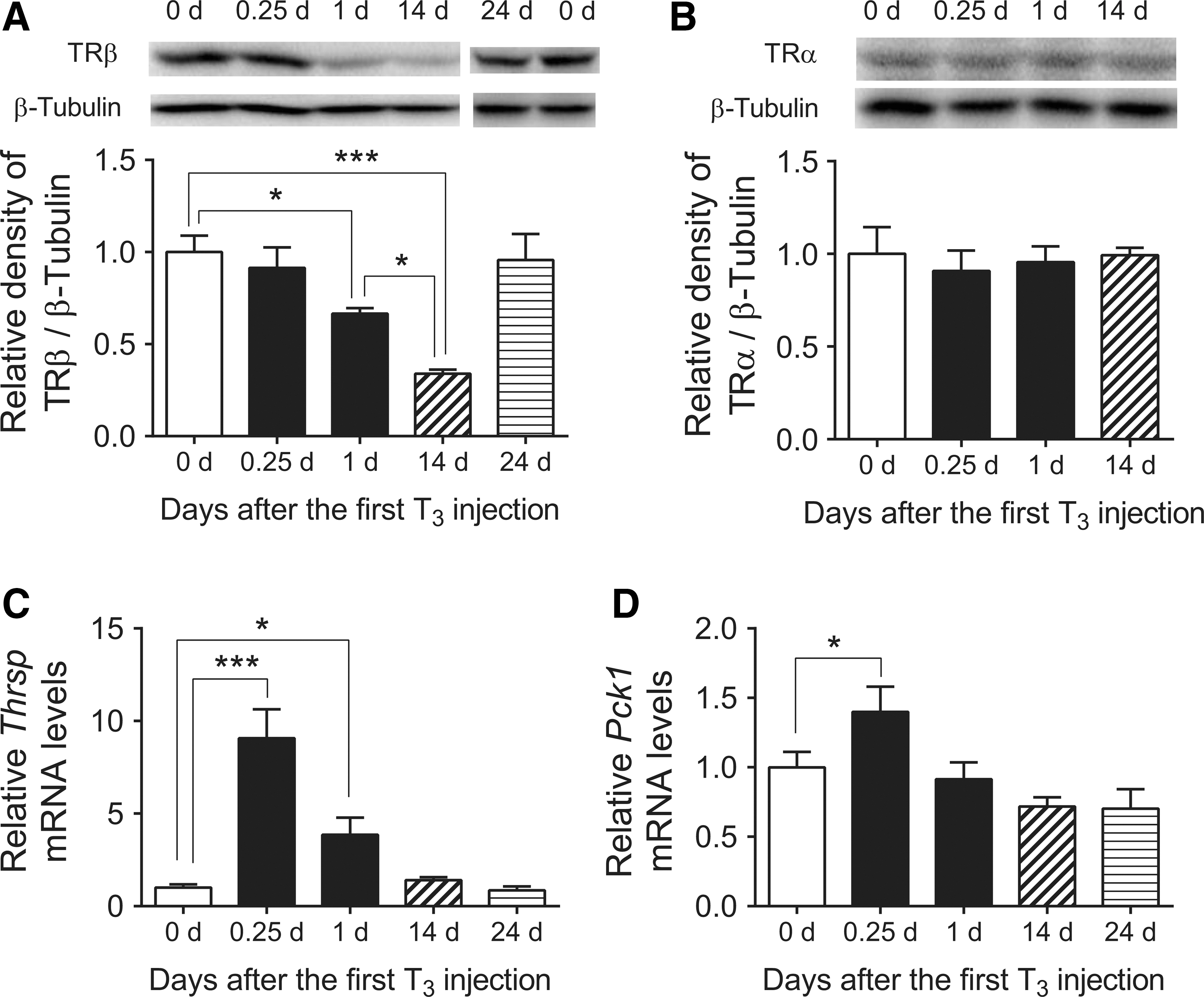
Expression of TRβ protein as well as Thrsp and Pck1 mRNA in mouse liver after T3 treatment. Adult male mice (8–10 weeks old) were treated with 20 μg of LT3/100 g of body weight for 14 days followed by cessation of LT3 for 10 days. (
In agreement with an earlier study (9), hepatic T3-target genes Thrsp and Pck1 mRNA expression became desensitized after chronic T3 treatment (Fig. 3C and D). To clarify further the possible contribution of decreased TRβ in expression of T3-target genes, THRB in THRB-HepG2 cells were knocked down using siRNA. Importantly, the amount of THRB knockdown was proportional to the decrease in PCK1 gene expression that was induced by T3 (Supplementary Fig. S3A and B). These data thus suggest that reduction in TRβ protein expression could play an important role in desensitizing T3-target genes.
Next, TRβ expression was examined in kidneys from mice that were chronically injected with T3. Surprisingly, no significant reductions in renal TRβ expression were found after chronic T3 treatment (Fig. 4A). In addition, renal Thrsp and Pck1 mRNA expression remained sensitive to chronic T3 exposure (Fig. 4B and C), whereas they became desensitized in the liver (Fig. 3C and D). These findings suggest that protein expression of TR isoforms and target gene expression are differentially regulated by chronic TH exposure in a tissue-specific manner.
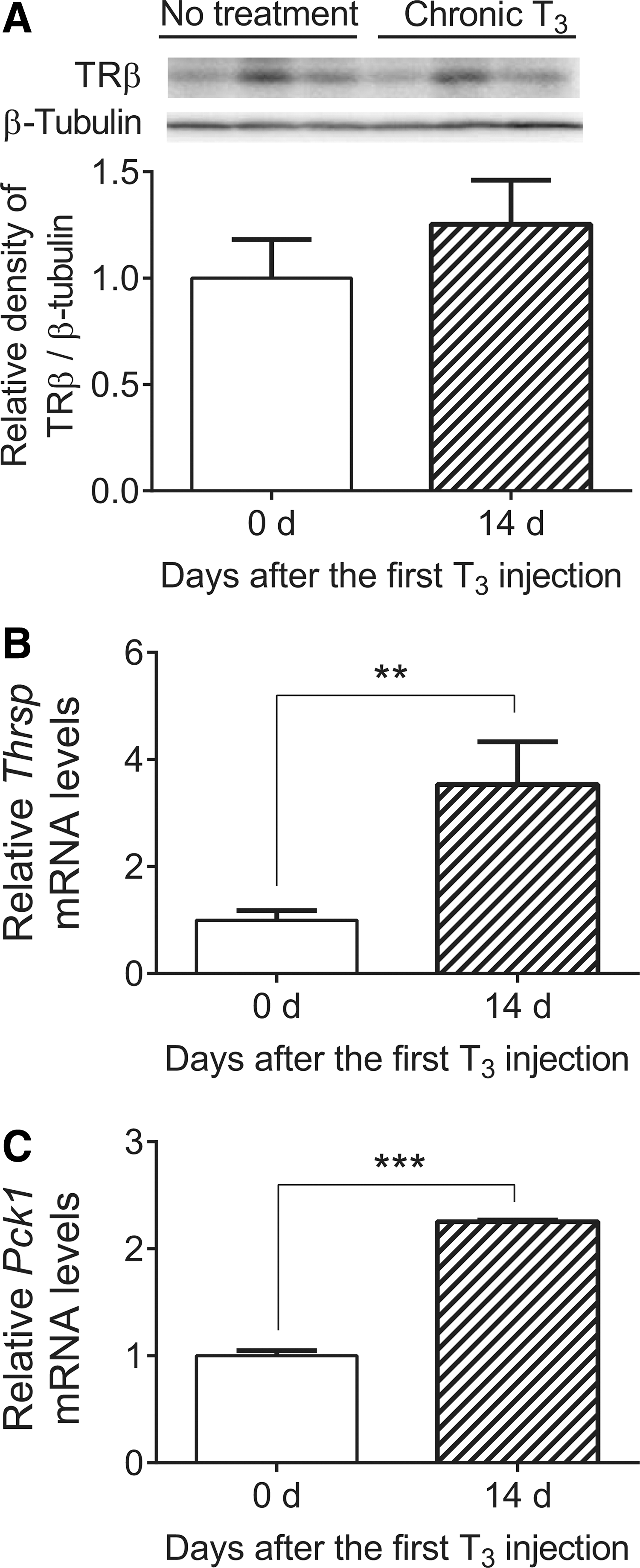
Expression of TRβ protein as well as Thrsp and Pck1 mRNA in kidney from mice treated with chronic T3. Adult male mice (8–10 weeks old) were treated with 20 μg of LT3/100 g of body weight for 14 days. Kidneys were harvested 14 days (chronic T3) after the first LT3 injection. (
In an earlier study, desensitization was confirmed by employing Mct8-knockout mice (9), an in vivo model of chronic intrahepatic hyperthyroidism since birth (14). Previous studies on adult Mct8-knockout mice showed that T3 levels were elevated not only in the liver but also in the kidney (15,16). In agreement with the findings observed in mice treated chronically with T3, TRβ expression was significantly decreased in the liver but not in the kidney in Mct8-knockout mice (Fig. 5A and B). Also, desensitization of a representative T3-target gene, Thrsp, was observed only in the liver (Fig. 5C and D). In addition, there were no significant changes in TRα, NCoR1, NCoA1, or RXRα protein expression in the liver between Wt and Mct8-knockout mice (Supplementary Fig. S4). Thus, there is a tissue
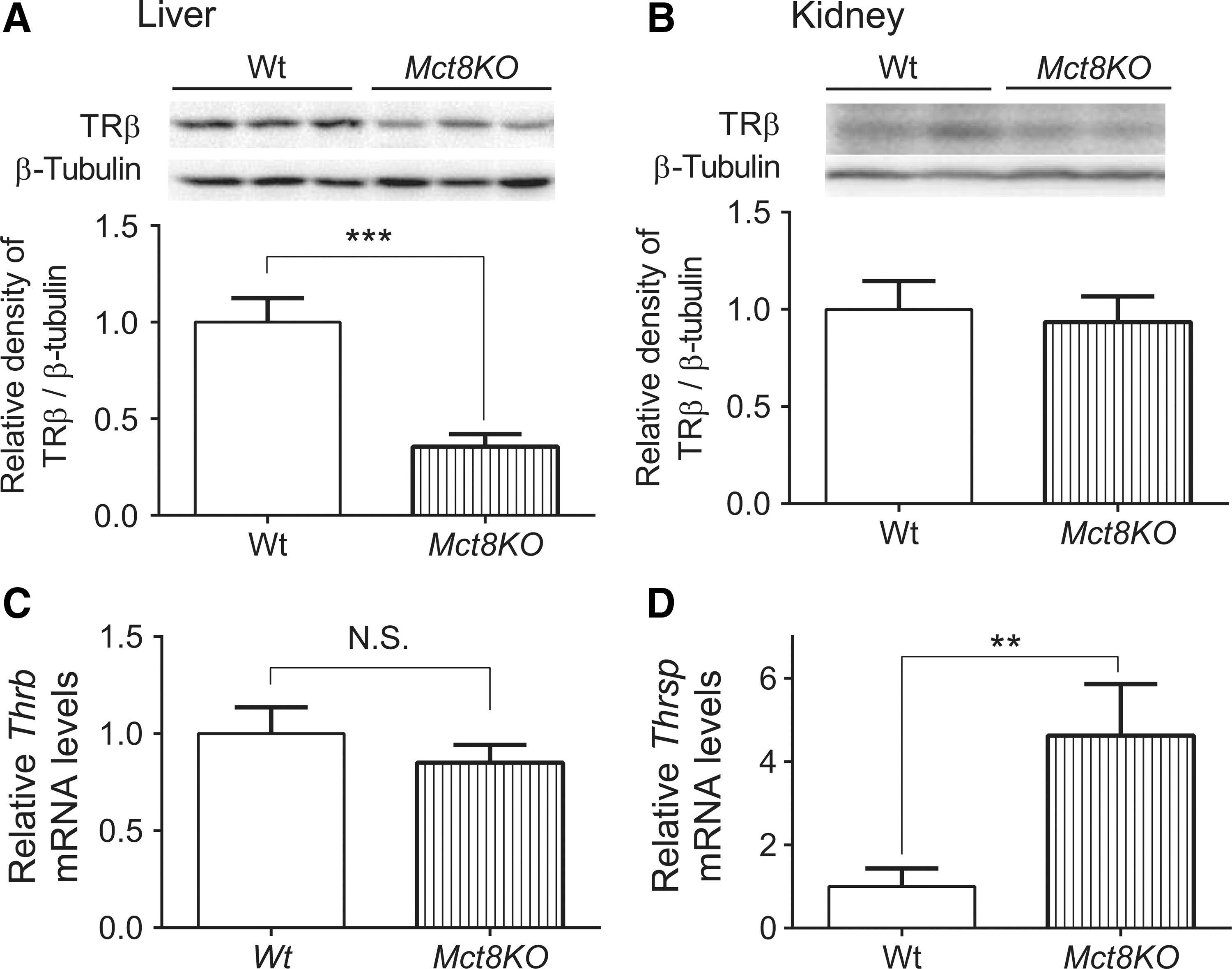
Expression of TRβ protein and Thrsp mRNA in the liver and kidney from Mct8-knockout (Mct8KO) mice. (
It was previously shown that hepatic long-chain acylcarnitine (LCAC) levels are increased after acute TH treatment (8). In contrast, there was no increase in LCAC levels after chronic T3 treatment. Moreover, surprisingly, significant increases in hepatic LCAC levels were observed after withdrawal (Fig. 6A). In this connection, pathway analyses of earlier microarray studies demonstrated that the lipid biosynthetic pathway (GO: 0008610; DAVID Bioinformatics Resources 6.7) was upregulated after acute T3 and T3 withdrawal but not during chronic T3 treatment (9). GO category 008610 includes well-established lipogenic genes, elongation of long-chain fatty acids family member 2 (Elovl2), Fasn, Elovl6, and fatty acid binding protein 5 (Fabp5).
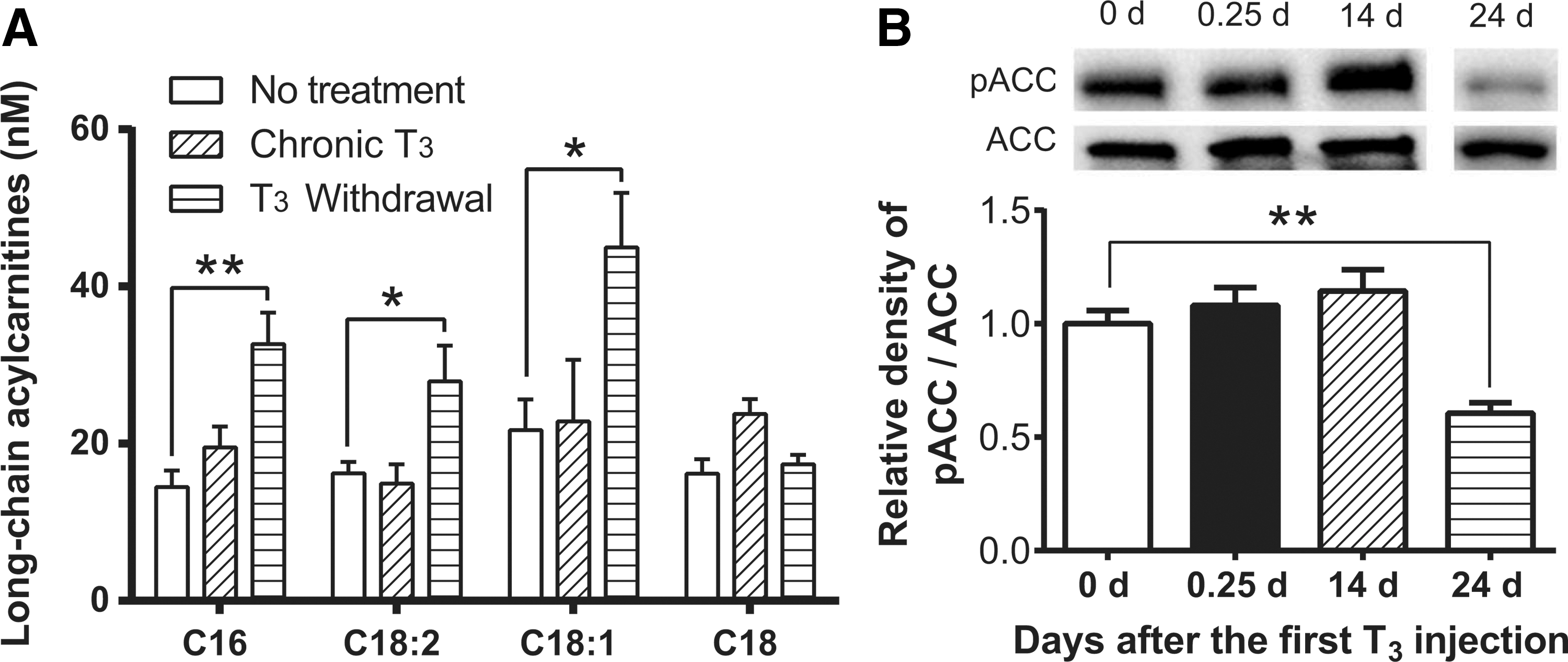
Long-chain acylcarnitines and phosphorylation of acetyl-CoA carboxylase (ACC) after T3 treatment and T3 withdrawal. Adult male mice (8–10 weeks old) were treated with 20 μg of LT3/100 g of body weight for 14 days followed by cessation of LT3 for 10 days. Livers were harvested 14 days (chronic T3) and 24 days after the first LT3 injection (T3 withdrawal). Metabolomics analysis of long-chain acylcarnitines (
To understand further lipid metabolism after T3 withdrawal, protein expression and phosphorylation of acetyl-CoA carboxylase (ACC) were analyzed. ACC plays an essential role in regulating fatty acid synthesis, and its phosphorylated form (pACC) is inactive. In agreement with metabolomics and microarray pathway analyses, T3 withdrawal decreased phosphorylation of ACC (Fig. 6B) in the liver, consistent with the occurrence of increased fatty acid biosynthesis during the transition from hyperthyroidism to euthyroidism. In addition, a trend toward increased triglyceride concentrations in the liver was found after T3 withdrawal (Supplementary Fig. S5). The hepatic levels of amino acids and organic acids were also analyzed after chronic T3 and T3 withdrawal (Supplementary Table S2). Interestingly, T3 withdrawal significantly increased hepatic lactate, an established lipogenic precursor (17).
Discussion
The present study shows that T3 decreases TRβ protein expression in a time-dependent manner (Fig. 3A), which could play a key role in transcriptional desensitization during chronic T3 exposure in the liver (Fig. 3C and D), since TRβ constitutes approximately 80–90% of total hepatic TRs in mice (18,19). Additionally, knockdown of THRB by siRNA resulted in decreased transactivation of PCK1 by T3 in human hepatocellular carcinoma cell line (Supplementary Fig. S3). Interestingly, even before the cloning and identification of TR isoforms, Samuels et al. reported that T3 treatment reduced the nuclear binding capacity of 125I-LT3 in a time- and dose-dependent manner in a rat pituitary cell line (20). A number of other studies subsequently revealed that TH regulated expression of TR isoforms both transcriptionally and post-transcriptionally in cell culture systems (21 –26). Also, alteration of TR expression has been thought to be an important mechanism controlling the tissue effects of TH under certain conditions, including non-thyroidal illness syndrome (27 –29). However, the long-term effects of TH on the mRNA and/or protein expression of TR isoforms or other regulators of TH action had not been previously examined in vivo. Accordingly, mRNA levels of TRs, TH transporters, Dios, RXRs, NCoRs, and NCoAs were determined in mouse liver, and only the two TR isoforms had decreased transcription after chronic T3 treatment (Fig. 2). Subsequent Western blotting analyses demonstrated that reductions in TRβ protein expression levels were more striking than changes in mRNA levels were (Fig. 3A), suggesting that post-transcriptional regulation plays a major role in the decreased TRβ protein expression. Additionally, this downregulation was reversible as TRβ protein levels returned to baseline when TH levels returned to normal after T3 withdrawal (Fig. 3A).
Several recent studies in cell culture models showed that T3-induced degradation of TRs was mediated by the ubiquitin-proteasome pathway, and may be essential for transcriptional regulation (23,25,26). For example, Dace et al. reported that transcriptional activity of TRβ was correlated with TRβ protein level in transfected CV1 cells. Moreover, proteosomal inhibition increased TRβ protein and T3-mediated transcriptional activity (23). Recently, Suh et al. reported that Sirtuin 1 promoted TRβ deacetylation in the presence of T3, which increased TRβ degradation in an ubiquitin-dependent manner (26). On the other hand, desensitization has been suggested as a possible adaptation against exposure to excessive TH concentrations. For example, decreased TRα expression was reported in the heart of mice with myocardial infarction (29), as well as in skeletal muscle from patients with non-thyroidal illness syndrome (28). To the best of the authors' knowledge, this is the first study to evaluate TRβ protein expression after acute versus chronic T3 treatment in vivo. Since the data suggest that post-transcriptional regulation plays a greater role in regulating TRβ expression (Figs. 2A and 3A), it is likely that proteosomal degradation of TRβ is an important mechanism for the transcriptional desensitization of target genes during chronic T3 exposure, and may be an adaptation against deleterious effects from chronic exposure to excessive TH.
Apart from desensitized target genes, previous microarray studies identified genes that were initially unresponsive but later became sensitive to T3 (e.g., Cyp17a1, Fgf21) as well as genes that were regulated both acutely and chronically (e.g., Dio1) (9). These findings demonstrate that a subset of hepatic target genes is regulated by chronic T3 treatment, despite decreased TRβ expression. A possible explanation for these effects is that TH indirectly regulates transcription of target genes by primary induction of transcription factors that regulate other genes. Furthermore, previous analysis of chromatin modifications demonstrated that increases in histone H3 acetylation at lysines 9 and 14 (H3K9/K14ac) and histone H3 trimethylation at lysine 4 (H3K4me3) may be important for transactivation during acute and chronic T3 treatments, respectively (9). Since the methylation of histones was reported to be a stable epigenetic mark (30), it is likely that H3K4me3 may contribute to persistent sensitivity of target genes during chronic T3 exposure despite decreased TRβ protein expression.
It has been reported previously that T3 regulates its target genes in a tissue-specific manner. For example, Cpt1a is responsive to thyroidal status in the liver but not in the heart (31). Also, mRNA expression of TR isoforms are differentially regulated by TH in a tissue-specific manner (21,24). Thus, TRβ expression was determined, as well as T3 responsiveness in the kidney, and they were compared with the liver after chronic T3. TRβ expression and induction of Thrsp and Pck1 was unaffected in the kidney. In contrast, in the liver, decreased TRβ expression and desensitization of Thrsp and Pck1 gene expression were observed after chronic T3 treatment (Figs. 3 and 4). These findings suggest that target genes in different tissues may be differentially regulated by chronic exposure to T3 due to their effects on TR expression.
This study also identified increased LCAC levels after T3 withdrawal but not after chronic T3 exposure, suggesting that metabolomic abnormalities could be associated with both desensitization and incomplete recovery observed for T3-target genes. Although TH has important roles in regulating hepatic metabolism, there have been very limited metabolomic studies of these effects. It was previously reported that acute T3 treatment induced LCACs (8). In this study, it was found that chronic T3 treatment no longer induced LCACs, whereas withdrawal increased LCACs above original baseline values (Fig. 6A). These findings correlate with the temporal expression pattern of lipogenic genes (i.e., Elovl2, Fasn, Elovl6, and Fabp5) identified by pathway analyses of earlier microarray studies (9). It is likely that the increased LCACs observed after T3 withdrawal were due, at least in part, to increased hepatic synthesis of fatty acids, although changes in circulating metabolites, especially free fatty acids, generated by lipolysis in white adipose tissue also may be contributory. A trend toward increased hepatic triglyceride content was also observed after T3 withdrawal. Varas et al. reported that changes in hepatic triglyceride accumulation were more prominent in acutely hyperthyroid than chronically hyperthyroid mice (32). The mechanism(s) for the increased hepatic LCAC levels and lipogenesis during recovery is not known but may be due to the transient hypothyroidism that occurred when there was increased TSH secretion by the pituitary but little endogenous thyroxine and T3 production by the thyroid (three days after withdrawal; day 17; Fig. 1), since the latter were still suppressed after chronic exposure to hyperthyroidism (9). Taken together, these findings suggest that desensitization and incomplete recovery of T3 target genes (particularly lipogenic genes) after chronic T3 and T3 withdrawal led to unexpected metabolomic changes that would not have been predicted based upon serum thyroid function tests alone.
In addition to observing increased LCACs after T3 withdrawal, decreased phosphorylation of ACC was found (Fig. 6B), suggesting that the lipogenesis pathway was indeed activated. Increased lactate is an established lipogenic precursor (17) and was increased in the liver. There also was a trend toward increased triglyceride concentration, consistent with the lipogenic pathway being activated functionally. Interestingly, a significant proportion of patients experience weight gain after successful therapy for hyperthyroidism (33). Although the precise mechanism(s) for this observation is not known, it has been suggested that a delayed adaptation of appetite and food intake in combination with the decreased energy expenditure after returning to euthyroidism may be contributory (34). However, it also is possible that increased lipogenesis after T3 withdrawal may contribute to weight gain during and after therapy in patients with hyperthyroidism.
In summary, this study has identified a time-dependent decrease in TRβ protein expression during chronic T3 treatment, which could play a key role in the transcriptional desensitization that occurs after chronic T3 exposure. The study also found that abnormalities in acylcarnitine levels were associated with desensitization during chronic T3 treatment and incomplete recovery observed for lipogenic genes after T3 withdrawal. Moreover, these findings suggest that circulating TH and TSH concentrations may not accurately reflect hepatic transcriptional and metabolic changes in some patients during chronic hyperthyroidism and/or recovery, and that careful clinical assessment of TH action in various target tissues is warranted. The identification and characterization of tissue-specific serum markers of TH action in the future may improve the diagnosis and treatment of these thyroid conditions.
Footnotes
Acknowledgments
We thank Kevin Fridianto, Sherwin Xie, Andrea Lim, Vania Lim, Patrick Lee, Dr. Benjamin Farah, Dr. Madhulika Tripathi, and Dr. Winifred Yau for technical assistance. This work was supported by the Singapore National Medical Research Council Clinician Scientist Award and Clinician Scientist Individual Research Grant 12May0004 (to P.M.Y.), by the Singapore Ministry of Health NMRC/BNIG/2025/2014 (to R.A.S.), by an Endocrine and Metabolic Society of Singapore grant (to M.K.-S.L.), by the A*STAR intramural funding for the Neuroepigenetics Laboratory under the Integrative Neuroscience Programme, Singapore Institute for Clinical Sciences (to J.C.G.S.), and by Grant R37-DK15070 from the National Institutes of Health (to S.R.).
Author Disclosure Statement
The authors declare that there are no conflicts of interest.