
Abstract
Background:
Thyroid hormones (THs) mediate pleiotropic cellular processes involved in metabolism, cellular proliferation, and differentiation. The intracellular hormonal environment can be tailored by the type 1 and 2 deiodinase enzymes D2 and D3, which catalyze TH activation and inactivation respectively. In many cellular systems, THs exert well-documented stimulatory or inhibitory effects on cell proliferation; however, the molecular mechanisms by which they control rates of cell cycle progression have not yet been entirely clarified. We previously showed that D3 depletion or TH treatment influences the proliferation and survival of basal cell carcinoma (BCC) cells. Surprisingly, we also found that BCC cells express not only sustained levels of D3 but also robust levels of D2. The aim of the present study was to dissect the contribution of D2 to TH metabolism in the BCC context, and to identify the molecular changes associated with cell proliferation and survival induced by TH and mediated by D2 and D3.
Methods:
We used the CRISPR/Cas9 technology to genetically deplete D2 and D3 in BCC cells and studied the consequences of depletion on cell cycle progression and on cell death. Cell cycle progression was analyzed by fluorescence activated cell sorting analysis of synchronized cells, and the apoptosis rate by annexin V incorporation.
Results:
Mechanistic investigations revealed that D2 inactivation accelerates cell cycle progression thereby enhancing the proportion of S-phase cells and cyclin D1 expression. Conversely, D3 mutagenesis drastically suppressed cell proliferation and enhanced apoptosis of BCC cells. Furthermore, the basal apoptotic rate was oppositely regulated in D2- and D3-depleted cells.
Conclusion:
Our results indicate that BCC cells constitute an example in which the TH signal is finely tuned by the concerted expression of opposite-acting deiodinases. The dual regulation of D2 and D3 expression plays a critical role in cell cycle progression and cell death by influencing cyclin D1-mediated entry into the G1-S phase. These findings reinforce the concept that TH is a potential therapeutic target in human BCC.
Introduction
T
Thyroid hormone has been reported to be a critical regulator of cell differentiation during development and in adulthood (4). Accordingly, the TH inactivating enzyme, D3, is overexpressed in several mouse and human cancers. This concept is exemplified by basal cell carcinoma (BCC), which is characterized by sustained D3 expression, and in which the TH signal attenuates cell proliferation thereby reducing BCC tumorigenicity (5). The skin is very sensitive to the action of TH. Indeed, TH influences both epidermal homeostasis and the pathogenesis of skin cancer (6). Moreover, normal TH levels are required for efficient epidermal homeostasis, function and regeneration (7). By binding to TRs, T3 regulates the expression of several keratins and skin-specific genes (8 –11). This regulation is critical for fetal epidermal differentiation, barrier formation, hair growth, sebum production, wound healing, epidermal oxygen consumption, and keratinocyte proliferation (8,12 –14).
The molecular mechanisms by which TH regulates rates of cell cycle progression and the relative contributions of deiodinases in this process have not yet been completely elucidated. We previously showed that D3 depletion and TH treatment influence the proliferation and survival of BCC cells. An intriguing and unexpected finding of the present study was that D2 is also highly expressed in BCC, which suggests that the D3-activating enzyme might play a role in BCC tumorigenesis and that the regulation of BCC tumorigenesis by TH can be more complex than previously thought.
The aim of the present study was to identify the molecular changes associated with cell cycle progression and apoptosis induced by TH and mediated by the deiodinases D2 and D3. Our data provide evidence that the BCC cell population is a unique cell model that is exquisitely sensitive to TH modulation and expresses high levels of both activating (D2) and inactivating (D3) TH enzymes. Co-expression of D2 and D3 raises the possibility of studying in detail the opposite effects of intracellular TH modulation and is thus a cellular model with which to unravel the molecular mechanisms of cell cycle control by TH.
Materials and Methods
Cell cultures, transfections, and reagents
BCC cells (G2N2c) are derived from transgenic mice expressing a constitutively active form of GLI2 under the control of the keratin 5 promoter (15), which generates BCC-like tumors called “trichoblastomas,” which are referred to as “BCC cells” throughout this article. Mouse primary keratinocytes were purified from wild-type (C57) mice and cultured under low-calcium conditions with 4% Ca2+-chelated fetal bovine serum and epidermal growth factor (Sigma-Aldrich, Saint Louis, MO) (16) Transient transfections were performed with Lipofectamine 2000 (Life Technologies Ltd., Paisley, Scotland) according to the manufacturer's instructions. Monoclonal anti-cyclin D1 anti-Erk antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA). BCC cells were treated with 2.5 μM and 10 μM doxorubicin for 16 hours and with 0.2 μg/mL and 1μg/mL tunicamicin for 24 hours.
Dio2 and Dio3 targeted mutagenesis
Depletion of Dio3 and Dio2 in mouse BCC cells was obtained using the CRISPR/Cas9 system from Santa Cruz Biotechnology (Dio3 CRISPR/Cas9 Knockout [KO] Plasmid, catalog sc-430733; Dio2 CRISPR/Cas9 KO Plasmid, catalog sc-420003) and control plasmid (Control CRISPR/Cas9 Plasmid, catalog sc-418922) as previously described (17). BCC cells were transfected with 500 ng of the D3, D2, or control CRISPR/Cas9 KO plasmids with Lipofectamine 2000 (Life Technologies). Three days after transfection, the cells were sorted using fluorescence activated cell sorting (FACS) for green fluorescent protein expression, single clones were analyzed by PCR to evaluate coding region alterations, and Dio2 exon 1 was sequenced to identify the inserted mutations. D3KO and control cells have already been checked for effective and specific D3 depletion as reported in Di Girolamo et al. (17).
Western blot analysis
Total protein extracts from BCC cells were run on a 10% SDS-PAGE gel and transferred onto an Immobilon-P transfer membrane (Millipore). The membrane was then blocked with 5% nonfat dry milk in phosphate-buffered saline (PBS), probed with anti-cyclin D1 and anti-PARP antibodies overnight at 4°C, washed, and incubated with horseradish peroxidase–conjugated anti-mouse immunoglobulin G secondary antibody (1:3,000), and detected by chemiluminescence (Millipore, cat. WBKLS0500). After extensive washing, the membrane was incubated with anti-Erk or anti-tubulin specific antibodies (Santa Cruz Biotechnology) as loading control. All Western blots were run in triplicate, and bands were quantified with ImageJ software.
Real-time PCR
Messenger RNAs were extracted with Trizol reagent (Life Technologies Ltd). Complementary DNAs were prepared with Vilo reverse transcriptase (Life Technologies Ltd.) as indicated by the manufacturer. The cDNAs were amplified by PCR in an iQ5 Multicolor Real Time Detector System (BioRad, Hercules, CA) with the fluorescent double-stranded DNA-binding dye SYBR Green (Applied Biosystems). Specific primers for each gene were designed to work under the same cycling conditions (95°C for 10 min followed by 40 cycles at 95°C for 15 s and 60°C for 1 min), thereby generating products of comparable sizes (about 200 bp for each amplification). Primer combinations were positioned whenever possible to span an exon–exon junction and the RNA digested with DNAse to avoid genomic DNA interference. Primer sequences are indicated in the Supplementary Table S1 (Supplementary Data are available online at
Colony formation assay
To evaluate colony formation, cells were seeded out in appropriate dilutions to form colonies. Three and six days after plating, cells were washed with PBS and stained with 1% crystal violet in 20% ethanol for 10 min at room temperature. Cells were washed with PBS twice and visible colonies were counted.
DNA transfection and Luciferase (Luc) expression assays
G2N2C cells were transiently transfected using Lipofectamine 2000 (Life Technologies Ltd.) according to the manufacturer's instructions. The reporter plasmids (pCcnD1CTR-Luc or TRE3-Tk-Luc) and CMV-Renilla were co-transfected into G2N2C. Luc activities were measured 48 hours after transfection with the Dual Luciferase Reporter Assay System (Promega), and differences in transfection efficiency were corrected relative to the level of Renilla Luciferase. Each construct was studied in triplicate in at least three separate transfection experiments.
Cell cycle analysis
For DNA content analysis, G2N2C cells were first synchronized by a double thymidine block protocol (18). Briefly, cells were grown in 100 mm plates to 40–50% confluence and then incubated for 14 h with growth medium supplemented with 2 mM thymidine. After two washes with PBS, cell medium was replaced by growth medium without thymidine for 24 h. Cells were then subjected to an additional incubation with 2 mM thymidine for 14 h followed by a release in growing medium until harvesting at the indicated time points. Cells were fixed in ice-cold 70% ethanol at −20°C. At least 10,000 cells were analyzed by FACS (FACS Canto2, Becton Dickinson) after staining with 5 μg/mL propidium iodide and exposure to 0.25 mg/mL RNase I (Sigma Aldrich). Data were analyzed with the MODFIT Lt3.0 Software.
For carboxy-fluorescein succinimidyl ester (CFSE) in vivo labeling, the CFSE labeling solution (1 μM) was added to 1 mL cell suspensions in PBS and incubated for 10 min at room temperature, after which cells were washed with regular medium to quench any free dye in solution. Cells were then plated in regular growing medium for different time points and harvested for cell sorting using a FACS Aria system (BD).
Cell synchronization measurements
Cells were synchronized through a double thymidine block (18). G2N2C cells were grown to 25–30% confluence, suspended in media containing 2 mM thymidine, and grown for 18 h (first block). Cells were then suspended with trypsin-EDTA (Life Technologies), divided into multiple containers, left to grow in normal growth media for 9 h, and then incubated for 17 h in media containing 2 mM thymidine (second block). Cells were released from the second block by incubation in growth media without thymidine and were harvested at the indicated time-points.
Statistical analysis
Differences between samples were assessed by Student's two-tailed t-test for independent samples; p-values <0.05 were considered significant. Relative mRNA levels (in which the first sample was arbitrarily set as one) are reported as the results of real-time PCR in which the expression of cyclophilin A served as housekeeping gene. All experiments were repeated and analyzed from three to five times.
Results
BCC cells express both the activating and inactivating thyroid hormone enzymes D2 and D3, respectively
We previously reported that BCC cells and tumors are characterized by elevated expression of the TH inactivating enzyme D3 (5). To probe in greater detail the mechanisms underlying TH metabolism in BCC cells, we analyzed the expression of other proteins related to TH metabolism, namely, D2, THRα, THRβ, MCT8, and MCT10, and compared their expression with that of normal mouse primary keratinocytes (Supplementary Fig. S1A). Surprisingly, BCC cells showed sustained expression of D2 mRNA; Furthermore, receptors and transporters were expressed at slightly lower levels in BCC cells than in mouse keratinocytes (Supplementary Fig. S1). Since sustained D2 expression was unexpected, we compared D2 expression in BCC cells with its expression in solid BCC and in known D2-expressing tissues, namely pituitary and brown adipose tissue, compared with a “low-expressing tissue,” namely, skeletal muscle. PCR analysis showed that BCC cells and tumors express consistent levels of D2, similar to unstimulated brown adipose tissue (Fig. S1B). These data demonstrate that BCC cells represent a complex TH-sensitive system with which to study the intracellular regulation of TH action.
D2 and D3 depletion inversely regulate intracellular TH availability
To address the functional relevance of D2 and D3 co-expression in BCC cells, we genetically depleted Dio2 (D2) or Dio3 (D3) gene expression using CRISPR/Cas9 technology. The generation of the CRISP-CTR and D3KO clones is reported elsewhere (17). D2 was depleted using the same CRISPR/Cas9 system used for D3-depletion (see methods section). Four different D2KO clones were selected, and effective D2 depletion was confirmed by DNA sequencing (Supplementary Fig. S2). To assess whether D2- and D3-depletion affect nuclear TH availability, we transfected the generated cells with a T3-dependent artificial promoter that drives the luciferase gene (TRE3-Tk-Luc promoter). As expected, TH activity was reduced in D2KO cells and enhanced in D3KO cells (Supplementary Fig. S3A). Interestingly, the T3 signal in D2KO cells was even lower than in normal BCC cells (which are already characterized by an “intracellular hypothyroid state”). This finding shows that D3 is not the only enzyme controlling the total thyroid signal within these cells. Similar results were obtained by measuring the expression of two TH-target genes in the skin, K10 and K14 (Supplementary Fig. S3B).
Targeted mutagenesis of D2 and D3 influences cell proliferation
To investigate and compare the functional consequences of genetic inactivation of D2 and D3 in BCC cells, we first evaluated cell proliferation in three clones for each condition (CTR, D2KO, and D3KO). Representative cell proliferation curves of one clone for each condition showed that D2KO cells grow faster than control cells, while D3-depletion in D3KO cells inhibits cell growth (Fig. 1A). EdU incorporation confirmed a growth advantage for the D2-depleted cells and a drastically decreased number of proliferative cells in a D3-null background (Fig. 1B). Similarly, colony assay demonstrated that D2-depletion accelerates cell proliferation, and that the opposite occurred in D3-depleted cells (Fig. 1C). These findings indicate that deiodinases D2 and D3 exert opposite effects on cell cycle control: D2-depletion accelerates cell proliferation, while D3-depletion inhibits cell replication.
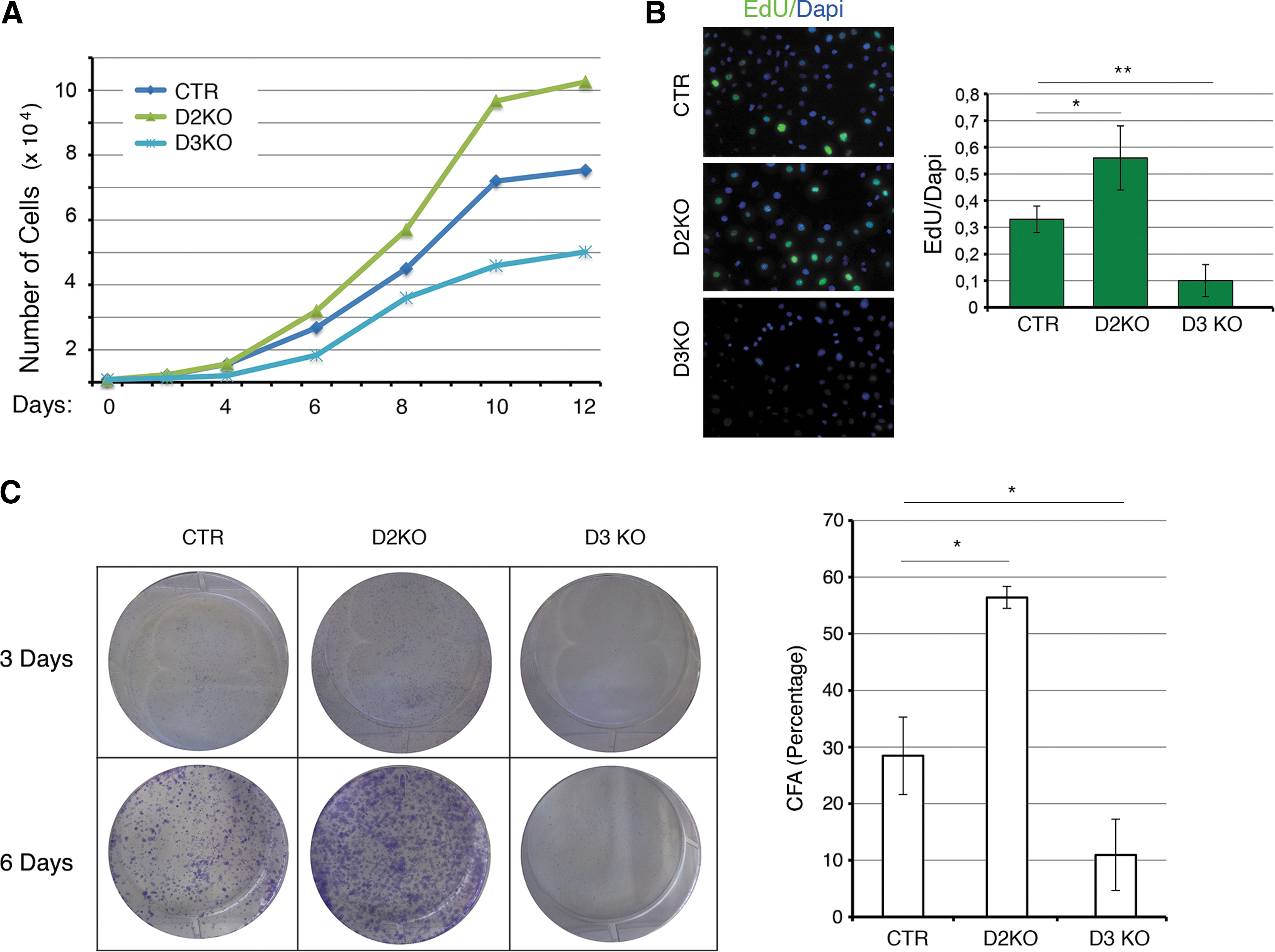
Selective type 2 and type 3 deiodinase (D2, D3) mutagenesis inversely regulates cell proliferation. (
We also treated D2KO clones with physiological T3 concentrations to assess if D2KO-dependent increases in cell proliferation were due to TH signal inactivation. T3 treatment decreased the proliferation rate of D2KO cells to almost that of CTR cells (Supplementary Fig. S4A). Analogously, we cultured D3KO cells in medium containing TH (normal serum) and in TH-deprived medium (charcoal stripped, CH). TH deprivation rapidly rescued the slow cycling profile of D3KO cells, which indicates that the reduced proliferation of D3KO cells depends on increased intracellular TH concentration (Supplementary Fig. S4B).
TH modulation by D2 and D3 affects cell cycle progression
We examined the cell cycle phase distribution in asynchronized cells using flow cytometry. D3 depletion decreased S-phase cells and increased the population of G1-phase and G2-phase cells (Fig. 2A); D2KO cells had a reduced number of G2-phase cells. To gain further insight into the effects of D2 and D3 depletion on cell cycle progression, we analyzed this process in S-phase synchronized cells by blocking the cell cycle with double-thymidine block and by forcing the cells to reenter the cell cycle by replacing thymidine in the medium. Cells were collected at different time points after thymidine removal. FACS analysis demonstrated that control cells recovered after thymidine block and that 37% of cells were in S-phase 6 hours after thymidine removal. Interestingly, D2-depleted cells divided more rapidly, and 73% of cells were in S-phase 6 hours after thymidine removal (Fig. 2B, Supplementary Fig. S5). On the contrary, the proportion of S-phase cells was much lower in the D3KO population (16%) than in control cells, with a consequent shift toward the G1 and G2 phases of the cell cycle. These data suggest that cell cycle progression of D3KO cells is drastically compromised. As a complementary approach, we used a fluorescence-based method for tracing the dividing cells. Cells were labeled with a fluorescent dye (CFSE) able to mark live cells so that when they divide, the daughter progeny receives approximately half the fluorescence of the parent. After labeling, cells were harvested at different time points to measure the dye incorporation by FACS analysis. Figure 2C shows that D2KO cells retained the dye for a shorter time compared with CTR cells, while D3KO cells were the longest retaining cells, thus implicating slower replication timing for these cells.
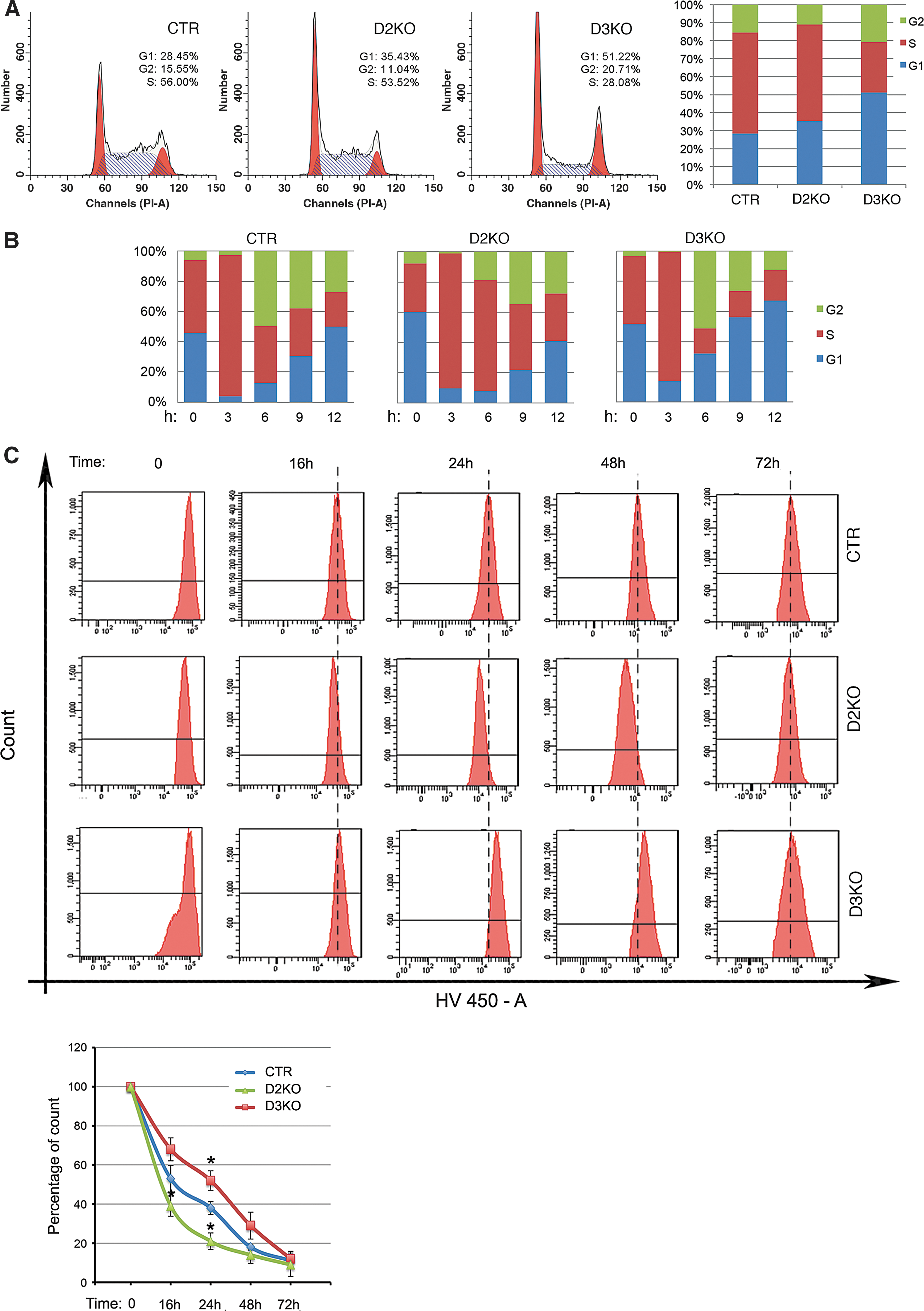
Cell cycle progression is significantly altered in D2- and D3-depleted cells. (
To determine the biochemical changes underlying these altered cell cycle profiles, we examined the cyclin D1 expression profile. Thymidine-treated cultures were released from thymidine block at different time points, and total lysates were analyzed for cyclin D1 by Western blotting (Fig. 3A, B). After synchronization, cyclin D1 increased more rapidly in D2KO cells than in control cells. Conversely, cyclin D1 levels were consistently very low in D3KO cells and increased slightly only nine hours after synchronization (Fig. 3A, B). Basal levels of cyclin D1 were lower in D3KO cells and higher in D2KO cells than in controls (Fig. 3C). A similar expression profile was observed at mRNA level (Fig. 3D). Taken together, these data indicate that concerted D2 and D3 action regulates S-phase entry and cyclin D1 expression.
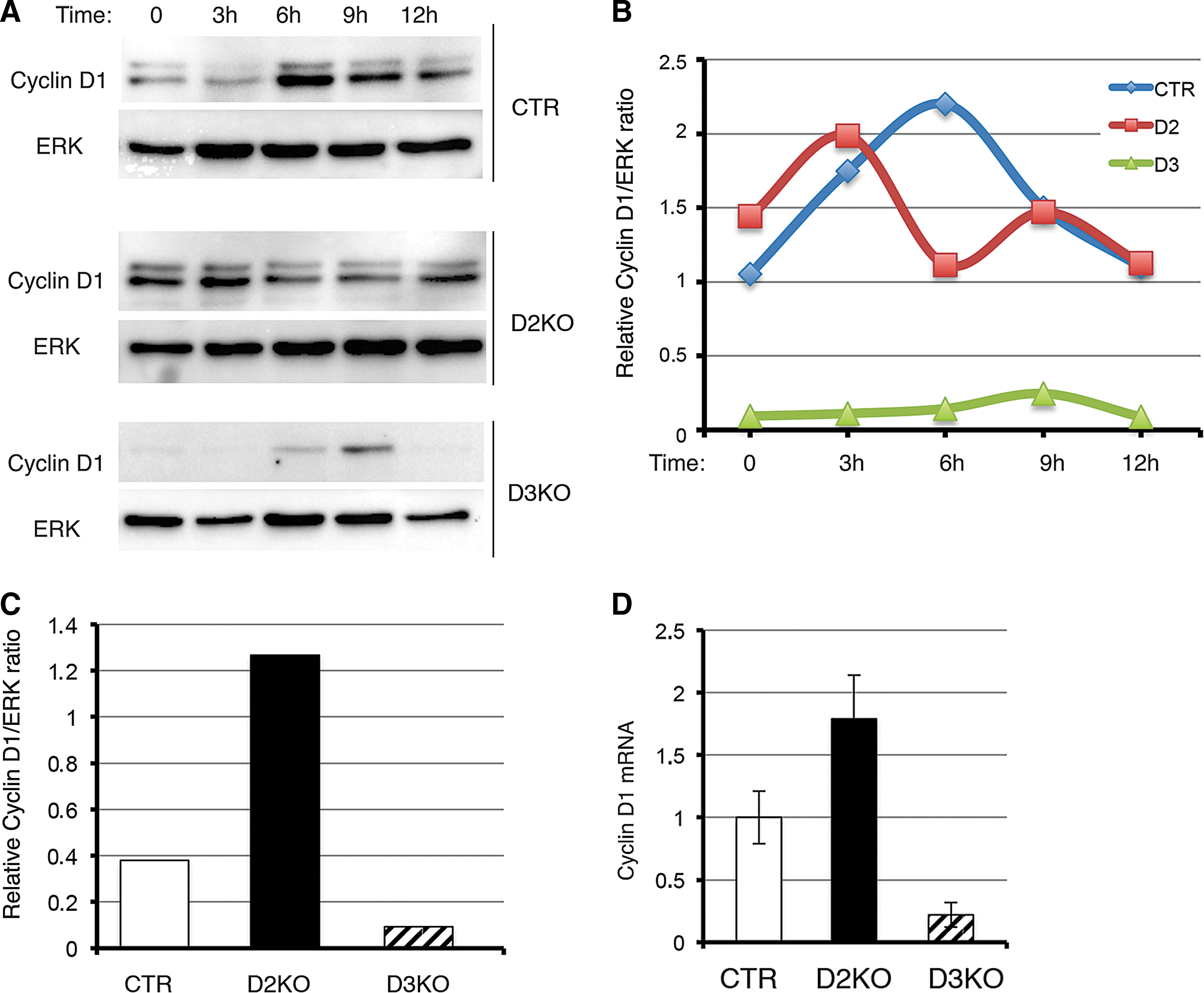
CyclinD1 expression reflects alterations in cell cycle in D2- and D3-depleted cells. (
D2-depletion protects BCC cells from cell death
Lastly, we analyzed the effects of D2 and D3 depletion on cell death. Annexin V incorporation and FACS analysis demonstrated that while apoptotic cells represent 4.4% of the control cell population, apoptosis was reduced to 1.5% in D2KO cells and increased to 14.8% in D3KO cells (Fig. 4A). TUNEL assay confirmed this result (Fig. 4B). PARP expression and cleavage confirmed that D2KO cells have a reduced basal apoptotic rate versus control cells (Fig. 4C). Interestingly, D2-depletion protected BCC cells also from doxorubicin- and tunicamicin- (data not shown) induced apoptosis, thus revealing a hitherto unrecognized role of hypothyroidism as an anti-apoptotic condition (Fig. 4D).
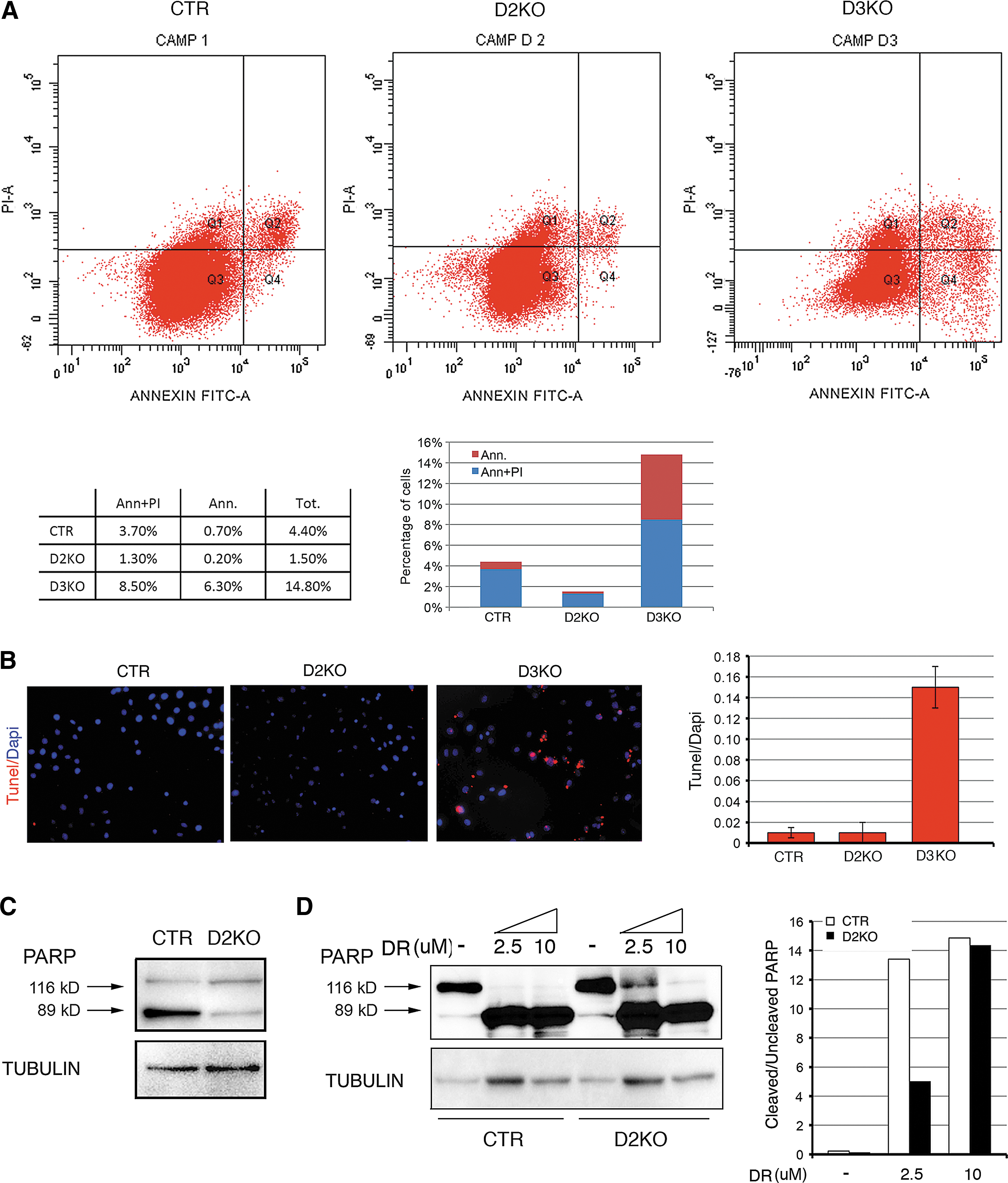
Elevated TH levels in D3KO cells enhance basal apoptosis. (
Discussion
The goal of this work was to dissect the role of deiodinases D2 and D3 in the regulation of intracellular TH signaling and their relative contribution to BCC cell tumorigenesis. Using a system of genetic depletion of the deiodinases D2 and D3, we generated D2KO and D3KO BCC cells, which have reduced and enhanced nuclear TH action respectively. The most prominent feature of these cells was a drastic alteration in cell proliferation rate, which reinforces the concept that TH plays a critical role in controlling cell proliferation. Given the pleiotropic roles played by TH in cell differentiation, growth, and metabolism, it is not surprising that the effects of TH on cell proliferation differ widely depending on the cellular context (19). Differential interaction with co-activator and co-factors, tissue-specific expression of different receptor isoforms, and the presence or absence of additional chromatin modifiers can explain the context-related effects of TH on cellular proliferation.
The BCC model is an example of high D3 expression within a tumor highly sensitive to TH (5,7,17). We have demonstrated that both treatments of BCC, with topical T3 or intracellular enhancement of the T3 signal by D3-depletion, affect BCC tumorigenesis by modulating multiple oncogenic signals, among them Shh/Gli2 (5) and miR21 (17). Consequently, we used the BCC cell line to unravel the molecular mechanisms by which TH interferes with the cell cycle machinery in a tumoral environment. Interestingly, while D3 was hitherto considered the deiodinase most frequently linked to tumoral transformation (19), we found robust levels of D2, the TH-activating enzyme, in BCC cells. Why is D2 expressed in BCC cells and what is its role in this tumoral context? We do not have a complete answer to this question, but the present study and our preliminary data indicate that BCC and colon cancer tumors (data not shown here) express D2 mRNA and protein besides D3, which implicates a possible role of D2 in tumorigenesis. Furthermore, other preliminary data on dual D2-D3 expression in BCC indicate that the two enzymes are partially co-expressed during tumor formation, with D3 being expressed in an early stage of tumorigenesis and D2 appearing thereafter (data not shown). It is conceivable that in some cellular contexts the presence of both activating and inactivating TH mechanisms is a means with which to rapidly customize the TH signal as necessary. The regulator of D2 expression in BCC and in the tumorigenesis in general is not known. Preliminary data from in vitro transfections in BCC cells indicate that none of the well-known inducers of BCC tumorigenesis (i.e., Gli2, β-catenin, and miR21) drive D2 expression in BCC. Consequently, further studies are required to clarify the regulation of D2 in vivo in BCC.
The most obvious phenotype of the generated deiodinase knockout cells was a drastic variation in cell proliferation. D2KO cells have low TH nuclear activity, as demonstrated by transactivation studies and the levels of TH target gene expression, and consequently represent a model of a “hypothyroid” condition. These cells are characterized by a high proliferation rate and by a high proportion of S-phase cells. Conversely, D3KO cells are slowly cycling cells and are characterized by increased nuclear TH activity when compared with control cells. These results point to a model in which TH slows the proliferation rate in the BCC context.
Studies in both synchronous and asynchronous cells indicate that variations of the proliferative rate induced by deiodinase depletion are associated to the proportion of S-phase and to cyclin D1 expression. Cyclin D1 is a key player in cell cycle modulation (20) and is dynamically regulated at both transcriptional and post-transcriptional level during the cell cycle to ensure the G1 to S progression of cells (21). Cyclin D1 is also a TH target in several cell contexts (19). However, the transcriptional regulation of cyclin D1 by TH is dynamic and highly variable depending on the context. Breast cancer is an example of a tumor in which the TH-TRβ complex reduces tumorigenesis by down-regulating cyclin D1 expression (22). Additionally, TRβ suppresses estrogen-dependent tumor growth of human breast cancer cells (23). Recently, it was suggested that the mechanism by which TRβ acts as tumor suppressor is via up-regulation of the nuclear receptor co-repressor 1 (NCOR1) and suppression of invasion, tumor growth, and metastasis in human hepatocarcinomas (24). Also in neuroblastoma cells, over-expression of TRβ inhibits the transcriptional response of the RAS/mitogen-activated protein kinase/ribosomal-S6 pathway, thereby suppressing growth (25). Similarly, overexpressed TRβ inhibited the AKT-mTOR-p70S6K pathway in human follicular thyroid cancer cells (26). Overexpression of cyclin D1 mRNA was detected in Thrβ PV/PV mice in which the Thrβ gene was mutated by an insertion leading to a frame-shift of TRβ1 and a total loss of triiodothyronine (T3) binding ability. The molecular mechanism by which this TRβ mutation increases cyclin D1 expression in Thrβ PV/PV mice was found to be due, at least in part, to the loss of the negative regulation of cyclinD1 promoter activity by wild-type receptor (27). On the other hand, there are many examples showing that TH and its receptors exert a positive effect on cell proliferation (28,29). Therefore, to better contextualize the transcriptional role of TH in cyclin D1 expression, we measured the levels of cyclin D1 mRNA and protein in synchronized BCC cells (5). Our data demonstrate that TH attenuation in D2KO cells potentiates the expression of this crucial cyclin, whereas the opposite occurs in D3KO cells.
Besides regulating cell cycle progression, modulation of T3 by D2 and D3 also affects BCC cell survival. Using our deiodinase loss-of-function BCC cell models, we evaluated the effects of T3 on apoptosis, and obtained evidence that T3 plays a pro-apoptotic role in BCC cells. Indeed, lower T3 levels in D2KO cells reduced basal apoptosis as measured by annexin V incorporation and FACS analysis, but they were also associated with stronger resistance to the apoptotic stimuli of doxorubicin and tunicamicin.
In conclusion, our results demonstrate that D2 and D3 are simultaneously expressed in BCC cells and that their action fine-tunes the TH signal. Manipulation of D2 or D3 dramatically affects the proliferative potential of BCC cells. Our results are compatible with the recent finding that precise regulation of the TH-dependent landscape of gene expression is essential in a vast range of biological processes, namely, embryonic development, tissue repair, and tumorigenesis (4). From a broad perspective, these novel concepts expand the use of deiodinases as a tool with which to interfere in diverse pathophysiological contexts in order to adapt the TH signal in a tissue-specific fashion.
Footnotes
Acknowledgments
This work was supported by grants from Associazione Italiana per la Ricerca sul Cancro to M.D. (IG 13065) and D.S. (IG 11362). We thank Jean Ann Gilder (Scientific Communication srl., Naples, Italy) for writing assistance.
C.M., M.A.D.S., D.D.G., E.D.C., A.G.C., and T.P. performed in vitro experiments and prepared figures; R.A. generated plasmids and performed in vitro experiments; M.R. and L.D.V performed fluorescence activated cell sorting analysis studies; D.S. provided observations, scientific interpretations, and contributed to experiment supervision and interpretation; M.D. designed the overall study, supervised the experiments, analyzed the results, and wrote the paper; and all authors discussed the results and provided input on the manuscript.
Author Disclosure Statement
The authors have declared that no conflict of interest exists.