
Abstract
Background:
Resistance to thyroid hormone alpha (RTHα), a disorder characterized by tissue-selective hypothyroidism and near-normal thyroid function tests due to thyroid receptor alpha gene mutations, is rare but probably under-recognized. This study sought to correlate the clinical characteristics and response to thyroxine (T4) therapy in two adolescent RTHα patients with the properties of the THRA mutation, affecting both TRα1 and TRα2 proteins, they harbored.
Methods:
Clinical, auxological, biochemical, and physiological parameters were assessed in each patient at baseline and after T4 therapy.
Results:
Heterozygous THRA mutations occurring de novo were identified in a 17-year-old male (patient P1; c.788C>T, p.A263V mutation) investigated for mild pubertal delay and in a 15-year-old male (patient P2; c.821T>C, p.L274P mutation) with short stature (0.4th centile), skeletal dysplasia, dysmorphic facies, and global developmental delay. Both individuals exhibited macrocephaly, delayed dentition, and constipation, together with a subnormal T4/triiodothyronine (T3) ratio, low reverse T3 levels, and mild anemia. When studied in vitro, A263V mutant TRα1 was transcriptionally impaired and inhibited the function of its wild-type counterpart at low (0.01–10 nM) T3 levels, with higher T3 concentrations (100 nM–1 μM) reversing dysfunction and such dominant negative inhibition. In contrast, L274P mutant TRα1 was transcriptionally inert, exerting significant dominant negative activity, only overcome with 10 μM of T3. Mirroring this, normal expression of KLF9, a TH-responsive target gene, was achieved in A263V mutation-containing peripheral blood mononuclear cells following 1 μM of T3 exposure, but with markedly reduced expression levels in L274P mutation-containing peripheral blood mononuclear cells, even with 10 μM of T3. Following T4 therapy, growth, body composition, dyspraxia, and constipation improved in P1, whereas growth retardation and constipation in P2 were unchanged. Neither A263V nor L274P mutations exhibited gain or loss of function in the TRα2 background, and no additional phenotype attributable to this was discerned.
Conclusions:
This study correlates a milder clinical phenotype and favorable response to T4 therapy in a RTHα patient (P1) with heterozygosity for mutant TRα1 exhibiting partial, T3-reversible, loss of function. In contrast, a more severe clinical phenotype refractory to hormone therapy was evident in another case (P2) associated with severe, virtually irreversible, dysfunction of mutant TRα1.
Introduction
R
While the identification of a number of RTHα cases has enabled key features of the disorder to be elucidated (10), how these characteristics are linked to the underlying THRA defect is ill-defined. Likewise, whether there is any relationship between the degree and reversibility of mutant TRα1 dysfunction in vitro and patient's response to thyroxine (T4) therapy is unclear. This article describes two adolescent males with de novo missense mutations (A263V and L274P) involving both TRα1 and α2 proteins. The divergent functional properties of these mutants, with lesser severity and greater reversibility of A263V mutant receptor dysfunction compared to L274P TRα1, correlates with differences in clinical characteristics of patients harboring the respective receptor defects and their responses to T4 therapy.
Patients and Methods
Case descriptions
Patient P1
P1 was the second child of healthy parents with one older sibling. Labor was induced at 39 + 4 weeks gestation due to large fetal size (birth weight 4.58 kg, 98th centile) and parturition complicated by shoulder dystocia. A small umbilical hernia was noted in the early postnatal period. The patient was breastfed for three months; weight was on the 98th centile from birth to at least two years of age, with length initially on the 91st centile (56.5 cm) but falling to the 9th centile (87 cm) at two and a half years. Bilateral inguinal hernia repair was performed in infancy (4 months and 10 months).
At eight months, maternal concern that he was excessively “placid” and with slow movements was supported by evidence of mild developmental delay. He only sat independently at eight months, walked at 24 months, and did not speak until after two years of age. In childhood, inability to use stairs, hop, use a ball, tie shoelaces (until 11 years of age), and cycle (until 12 years age of age, requiring specific tuition for this) suggested problems with coordination. Objective assessment (at nine years of age) confirmed poor motor skills (<5th centile for age) and an unspecified developmental coordination disorder for which he received occupational therapy. Several persistent deciduous teeth (removed at 15 years of age) indicated delayed dentition.
Referral to a pediatrician for possible delayed puberty at 16 years of age (although he was in mid-puberty G4P3, testicular volume 20 mL, when assessed) documented an unusual pattern of thyroid function tests (thyrotropin [TSH] 3.20 mIU/L [reference range 0.27–4.20 mIU/L], free T4 [fT4] 9.0 pmol/L [reference range 12–22 pmol/L], free T3 [fT3] 8.0 pmol/L [reference range 3.9–6.7 pmol/L]), prompting further investigation. With a little teaching support, he has completed secondary-level education and is currently enrolled at university.
Patient P2
P2 is the fourth child of healthy parents and siblings. Following a normal pregnancy, labor was induced at 41 + 5 weeks. He weighed 3.26 kg (9th centile), and coronal hypospadias and umbilical hernia was noted. Failure to thrive in the first six months of infancy (weight <0.4th centile) necessitated energy-dense formula feeding. Developmental delay, first noted at nine weeks, persisted throughout childhood. He first sat at 12 months, pulled himself to stand at 22 months, and walked at two and a half years. Formal assessment at two and a half years of age showed delayed gross motor skills (at 12-month level) and significant speech delay (only two-word vocabulary). Other notable features included dysmorphic facies, truncal hypotonia, delayed dentition (first tooth eruption at two years of age), myopic astigmatism, genu valgum, ankyloglossia, skeletal dysplasia (Wormian bones, short tubular hand bones, delayed closure of the anterior fontanelle, coxa valga, femoral epiphyseal dysgenesis, mesomelic shortening of upper and lower limbs, delayed bone age [bone age one year and three months at chronological age three years and three months]), bilateral hydroceles, and constipation. Abnormal thyroid function tests presumed indicative of central hypothyroidism (TSH 2.4 mIU/L [reference range 0.4–5.5 mIU/L], fT4 9.0 pmol/L [reference range 10–18.7 pmol/L]) at 11 years of age prompted commencement of T4 treatment. Subsequent pubertal development was normal. He attends a school for children with special needs and has persistent learning disability. Abnormal thyroid function and global developmental delay prompted further investigation at 15 years of age.
Methods
All investigations were undertaken either as part of a protocol approved by a local Research Ethics Committee (Cambridgeshire, LREC 98/154) and/or were clinically indicated and undertaken with prior written informed consent of patients and/or parents.
Biochemical measurements
Thyroid function tests (fT4, fT3, TSH, reverse T3 [rT3]) and biochemical measurements (creatine kinase [CK], sex hormone binding globulin, IGF1, lipids, hematocrit) were assayed, as described previously (6).
Resting energy expenditure and body composition
Resting energy expenditure (REE) was measured by indirect calorimetry using a ventilated canopy (GEM; GEMNutrition, Daresbury, United Kingdom) and body composition using dual energy X-ray densitometry (DEXA; Encore; GE Medical Systems, Madison, WI). The coefficient of variation for repeated REE measurement in subjects is +4% (0.24 MJ/day).
Molecular genetic studies
Coding exons of the THRA gene were polymerase chain reaction (PCR) amplified from genomic DNA using specific primers and analyzed by Sanger sequencing, as described previously (1).
Construction of wild-type, A263V, or L274P mutant TRα and other vectors
Receptor mutations were generated by site directed mutagenesis of wild-type (WT) human TRα1 or TRα2 cDNAs, and cloned into a eukaryotic expression vector (pcDNA3), downstream of a Flag epitope tag. Full length WT and mutant TRα1 cDNAs were cloned in pCMX-VP16 (kind gift from R. Evans) to yield VP16-TRα1 fusions. Constructs expressing the Gal4DBD alone or fused to receptor-interacting domains of NCoR and TRAP220 and reporter genes containing a thyroid response element (MALTKLUC) or GAL4 binding site (UASTKLUC) have been described previously (6).
Electrophoretic mobility shift assays
32P-labeled probes of a direct repeat thyroid response element from the malic enzyme gene were incubated with in vitro translated WT and mutant TRα1 proteins with or without RXR and 100 nM of T3 and analyzed by gel electrophoresis, as described previously (6).
Transfection assays and Western blotting
For transcriptional activity and dominant negative inhibition assays, JEG-3 cells, cultured in 96-well plates with OptiMEM/10% stripped fetal calf serum, were cotransfected with MALTKLUC, TR expression vectors, and a plasmid (Bos-β-gal) to monitor transfection efficiency, assaying luciferase and beta-galactosidase activity at 36 hours following T3 exposure, as reported previously (6). Data shown are normalized to maximal reporter gene activation with WT receptor and represent the mean and standard error of the mean (SEM) of at least five independent experiments, each performed in triplicate. In two hybrid assays, JEG-3 cells were cotransfected with UASTKLUC, Gal4DBD- and VP16-TR fusion constructs, and Bos-β-gal, assaying luciferase and beta-galactosidase activity as above. Values shown are expressed as a percentage of the WT maximum response and are the mean and SEM of at least five independent experiments each undertaken in triplicate. Western blotting of JEG-3 cell lysates using Flag (F3165; Sigma–Aldrich, St. Louis, MO) and VP16 (sc-7545; Santa Cruz Biotechnology, Dallas, TX) antibodies quantitated TR and VP16 fusion proteins, respectively, with beta-actin levels verifying comparable protein input.
Quantitative real-time PCR analysis of gene expression
Peripheral blood mononuclear cells (PBMCs) were isolated by Ficoll gradient centrifugation and cultured for 24 h in RPMI/5% stripped fetal bovine serum in the presence of increasing concentrations of T3. RNA, isolated using a RNeasy Mini Kit (Qiagen, Hilden, Germany) was reverse transcribed and analyzed by Taqman quantitative real-time PCR (qPCR), as described (6), using KLF9 (Hs00230918_m1) and 36B4 Taqman probes (Applied Biosystems, Foster City, CA). The comparative Ct method was used to quantify transcripts and normalize to 36B4 expression levels, which did not vary with ligand treatment.
Crystallographic modeling
Models of A263V or L274P mutant TRα1 were based on structures of WT TRα1 (3ILZ) and generated using the MacPyMOL Molecular Graphics System, v1.5.0.4 (Schrödinger, LLC, New York, NY).
Results
Clinical investigation
Patient P1 is of average proportionate height (50th centile; subischial leg length +1 SD; Fig. 1A and Table 1). He has mild macroglossia and slightly broad facies with a few skin tags (Fig. 1B). His head circumference is increased (62.5 cm; >99th centile); a skull radiograph showed a thickened calvarium (Fig. 2A) and delayed dentition (Fig. 2C). Abdominal and pelvic imaging showed a loaded bowel (Fig. 2B) with normal femoral epiphyses (Supplementary Fig. S1; Supplementary Data are available online at
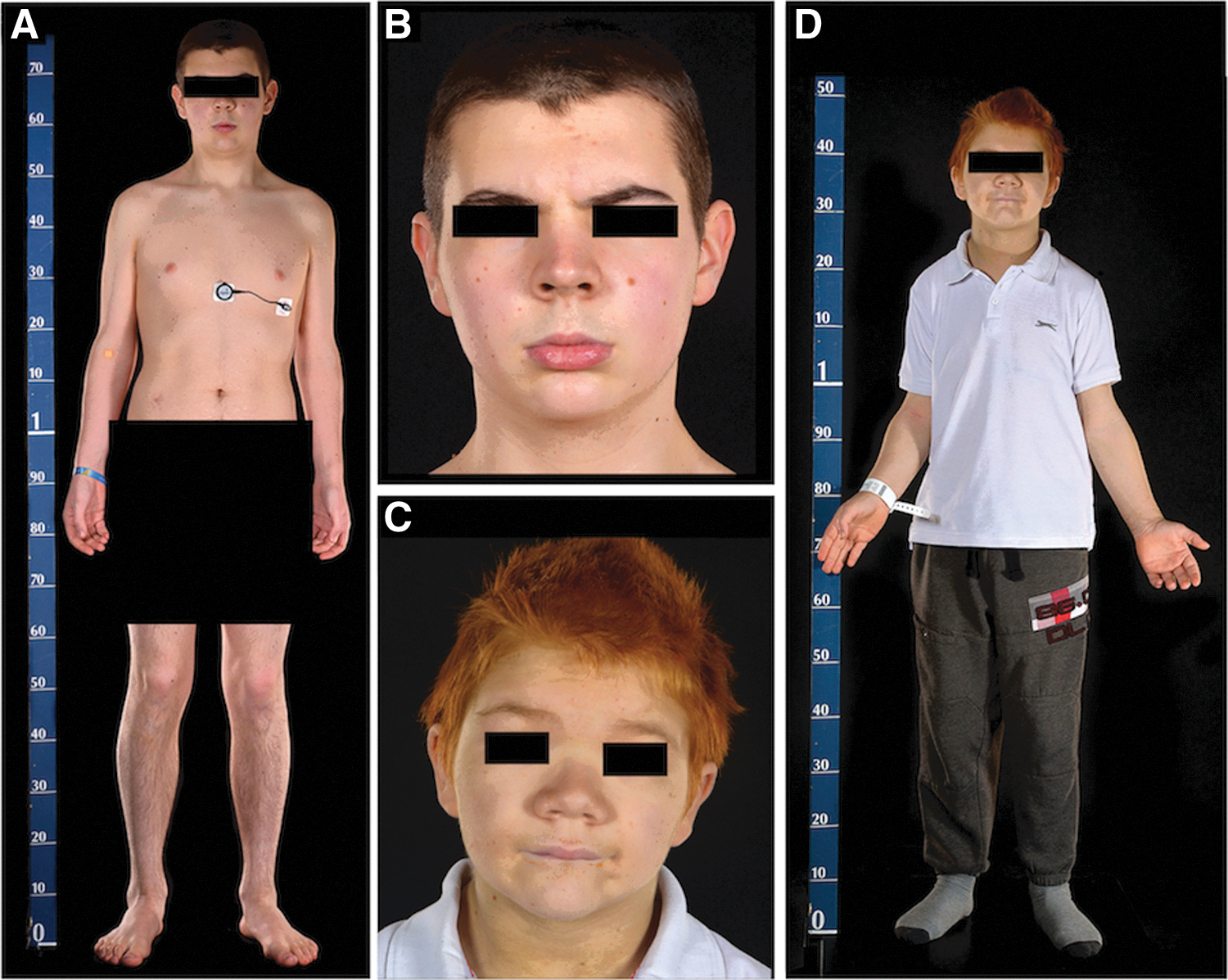
Phenotypic characteristics of patient P1 (
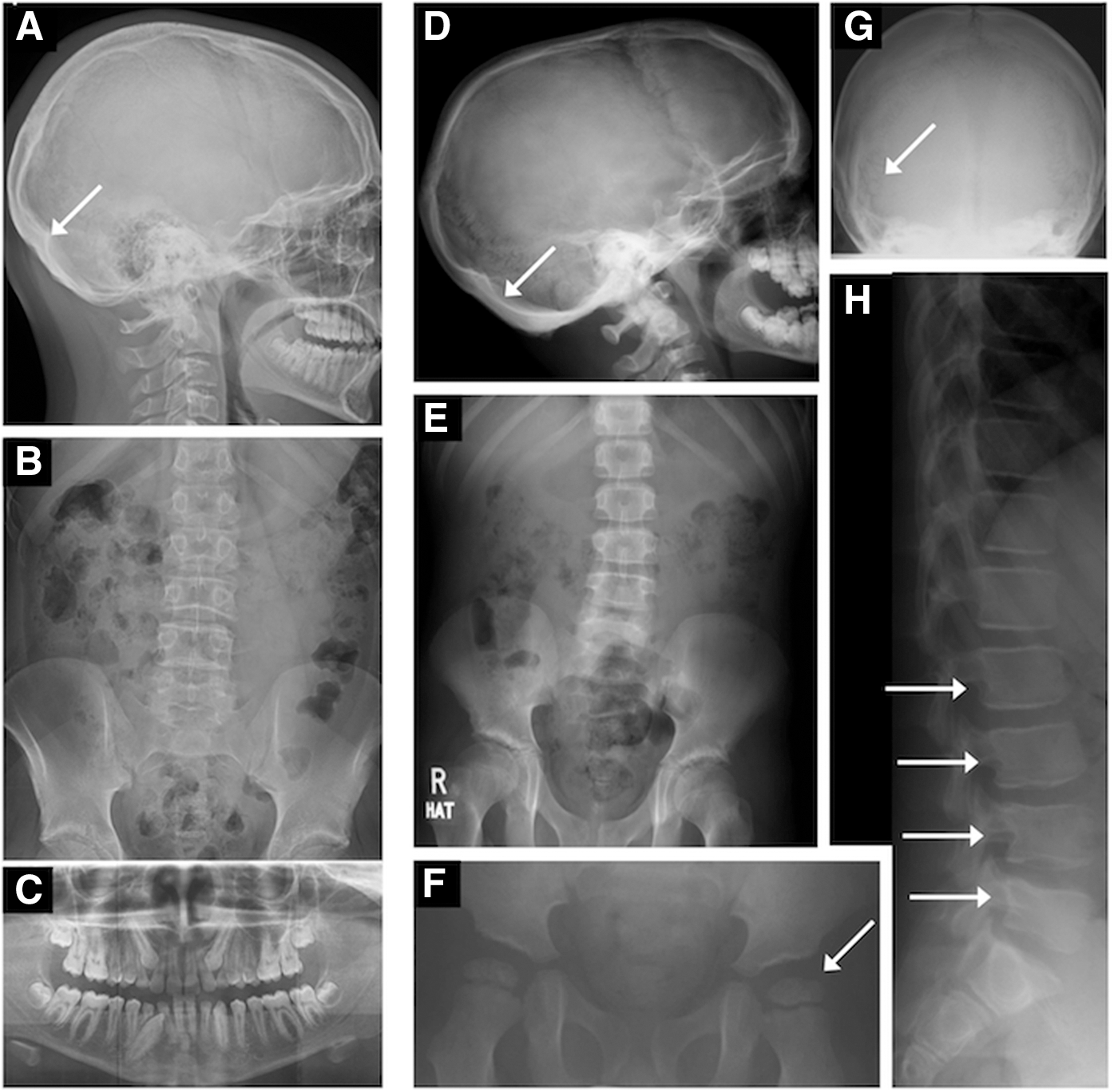
Radiological abnormalities in patients P1 and P2 illustrating thickened calvarium in P1 (
+ Indicates present, mild; ++ indicates present, moderate; +++ indicates present, severe/marked.
Post T4 therapy.
In childhood.
T4, thyroxine; T3, triiodothyronine.
For some parameters (resting energy expenditure, sleeping heart rate, SHBG), reference values (mean and range) from age- and sex-matched healthy children are shown.
An interim REE measurement in P1, at 17.5 years of age on 75 μg T4 therapy, was 0.133 MJ/kg lean body mass/day.
REE, resting energy expenditure; SHR, sleeping heart rate; TSH, thyrotropin; fT4, free T4; fT3, free T3; rT3, reverse T3; CK, creatine kinase; SHBG, sex hormone binding globulin; LDL, low-density lipoprotein.
Patient P2 has disproportionate short stature (height 0.4th centile; normal sitting height with subischial leg length −2 SD; Fig. 1D and Table 1). He has marked macroglossia and is dysmorphic with broad facies, marked hypertelorism, flat nasal bridge, puffy skin, and skin tags (Fig. 1C). His head circumference is increased (60.5 cm; 99th centile); a skull radiograph at four years of age showed multiple Wormian bones (Fig. 2G), and his calvarium is now thickened (Fig. 2D). Femoral epiphyseal dysgenesis seen in early childhood (Fig. 2F) is no longer evident (Fig. 2E). Abdominal imaging also showed a loaded bowel (Fig. 2E), and a spine radiograph showed posterior scalloping of the lumbar vertebrae (Fig. 2H). Prior to treatment, his fT4 was subnormal with raised fT3 and low rT3 levels. Although his resting energy expenditure was normal, muscle CK levels were raised (Table 1).
Molecular genetic studies
THRA sequencing indicated that both patients were heterozygous for missense mutations (P1:c.788C>T, p.A263V; P2:c.821T>C, p.L274P) involving both TRα1 and α2 proteins. In both cases, the mutations occurred de novo, with neither parent being affected. In each proband, the mutations were present in DNA isolated from different cell types (PBMCs, buccal epithelial tissue, hair follicle; Supplementary Figs. S2 and S3) and are not present in genome databases (dBSNP, Exome Aggregation Consortium (ExAC), Cambridge, MA;
Functional properties of A263V and L274P mutant TRα1 and α2 proteins
In functional assays, hormone-dependent transcriptional activation by A263V mutant TRα1 is impaired at low T3 concentrations (0.01–100 nM), but is comparable to WT receptor at higher T3 levels (1–10 μM). In contrast, L274P mutant TRα1 exhibits negligible transcriptional activity except at the highest T3 concentrations (10 μM; Fig. 3A). Consistent with this, in protein–protein interaction assays, A263V mutant TRα1 dissociates from corepressor (CoR) and recruits coactivator (CoA) at lower T3 concentrations (CoR dissociation 100 nM; CoA recruitment 1 μM) than L274P mutant TRα1 (maximal CoR dissociation and CoA recruitment at 10 μM; Fig. 3C and D).
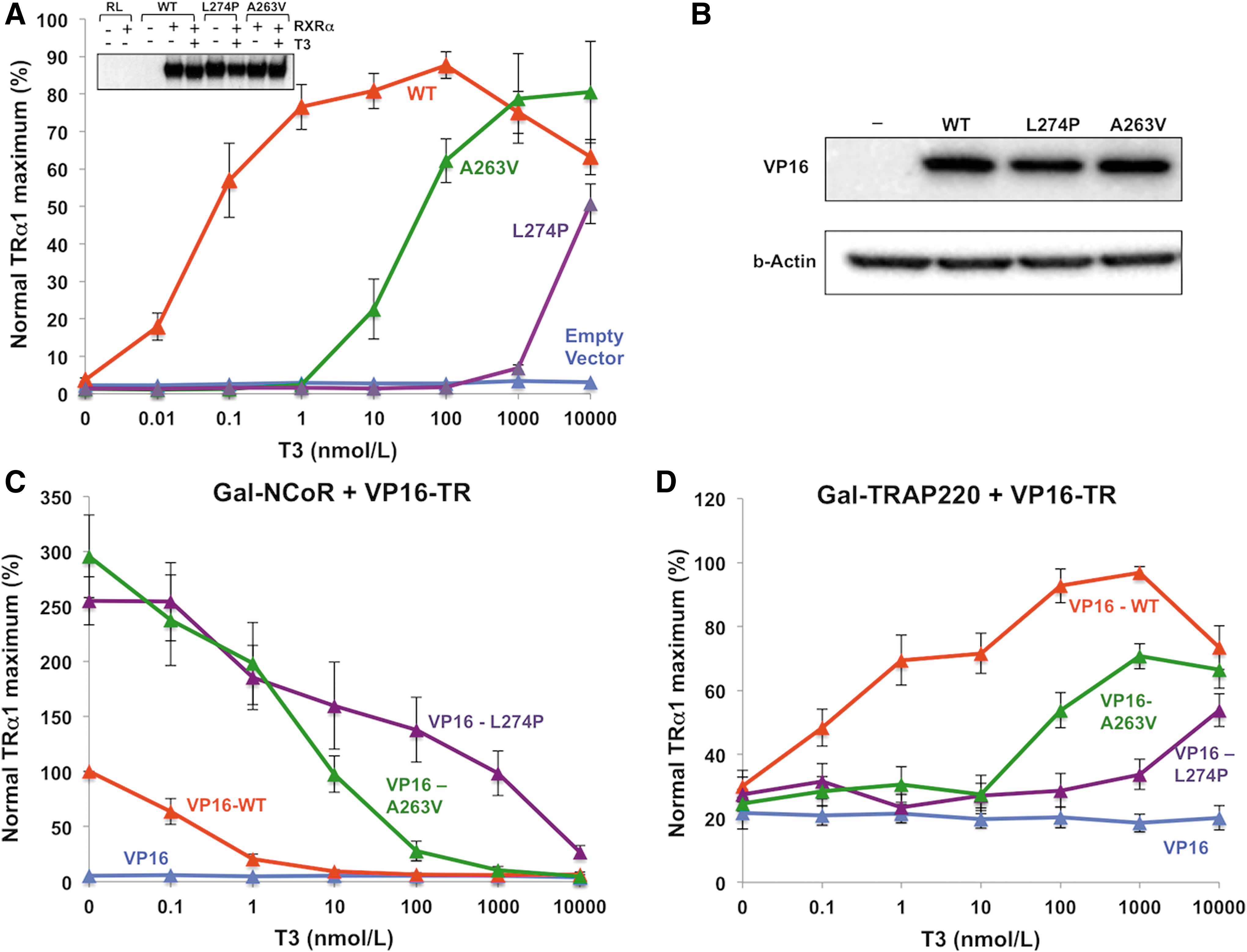
Functional properties of A263V and L274P mutant thyroid hormone receptor α1 (TRα1). (
When coexpressed, mutant receptors inhibit the transcriptional activity of their WT counterparts in a dominant negative manner. Lower T3 levels (100 nM) reverse such dominant negative inhibition by A263V mutant TRα1 than inhibition by L274P mutant TRα1 (10 μM; Fig. 4A). Correlating with this, reduced expression of a known TH-responsive target gene (KLF9) is normalized by exposure of A263V mutant TRα1-containing PBMCs to lower concentrations of T3 (1 μM), with L274P mutant TRα1-containing cells being refractory, even with 10 μM of T3 exposure (Fig. 4B).
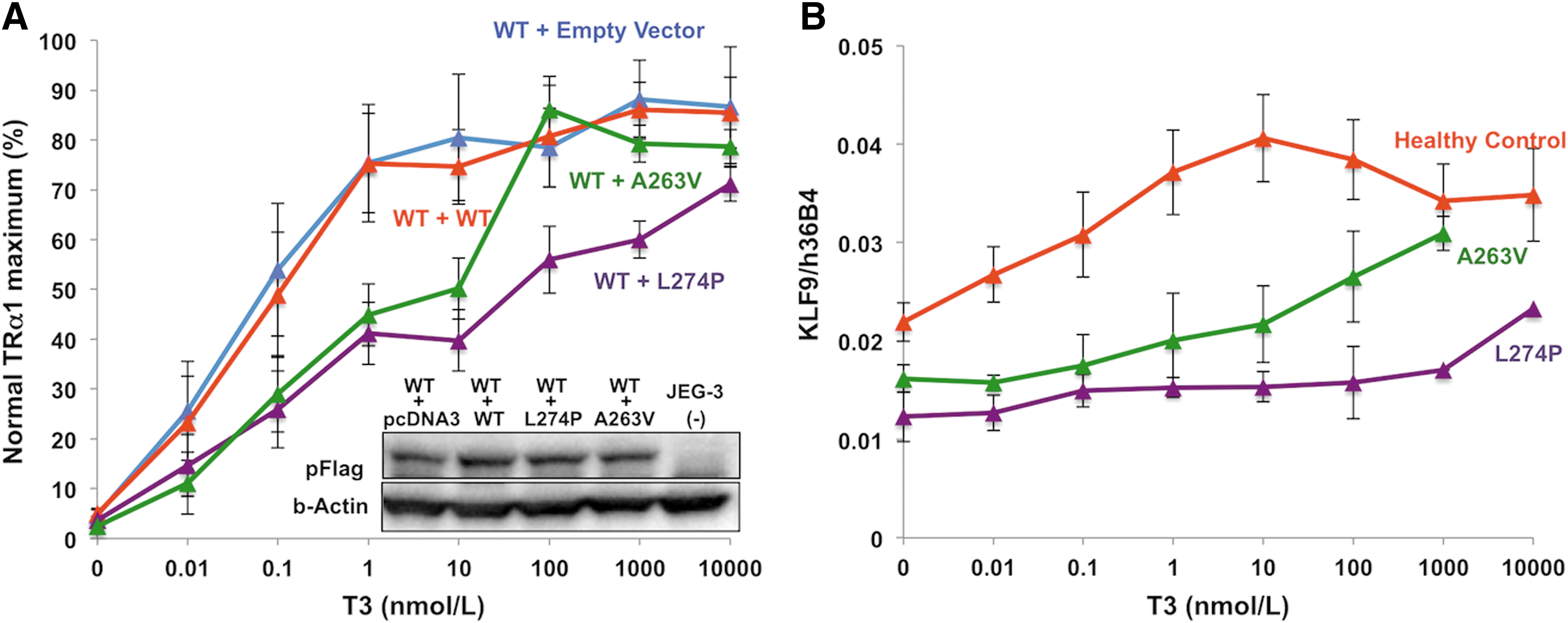
(
When studied in the TRα2 background, the L274P mutant α2 was transcriptionally inert, similar to WT α2 (Supplementary Fig S4A). As with WT TRα2, it inhibited the function of WT TRα1, but only when coexpressed at 10- to 50-fold higher levels (Supplementary Fig. S4B). These properties are wholly analogous to those of A263V mutant α2, which have been reported previously (6).
Response to T4 therapy
T4 therapy for 18 months in patient P1 (A263V mutant TRα), accompanied by an appropriate rise in fT4, fT3, and rT3 levels (Table 2), was associated with improved linear growth (Fig. 4, left panel) and striking reduction in weight (Fig 4, right panel), largely due to diminished fat mass (pretreatment: total body fat mass 26,950 g, 31%; posttreatment: total body fat mass 20,520 g, 23%). Significantly, such an improvement in body composition was associated with beneficial metabolic changes, with a reduction in fasting insulin and a rise in circulating adiponectin levels (pretreatment: insulin 156 pmol/L [reference range 0–60 pmol/L], adiponectin 3.4 μg/mL; posttreatment: insulin 75 pmol/L, adiponectin 7.1 μg/mL). In contrast, despite T4 treatment since 11 years of age, achieving similar changes in thyroid hormone levels (Table 2), patient P2 (L274P mutant TRα) exhibited negligible change in linear growth (Fig. 5 left panel) and ongoing weight gain, which was only reversed when on a higher T4 dose (62.5 μg) from 14 years of age (Fig. 5, right panel).
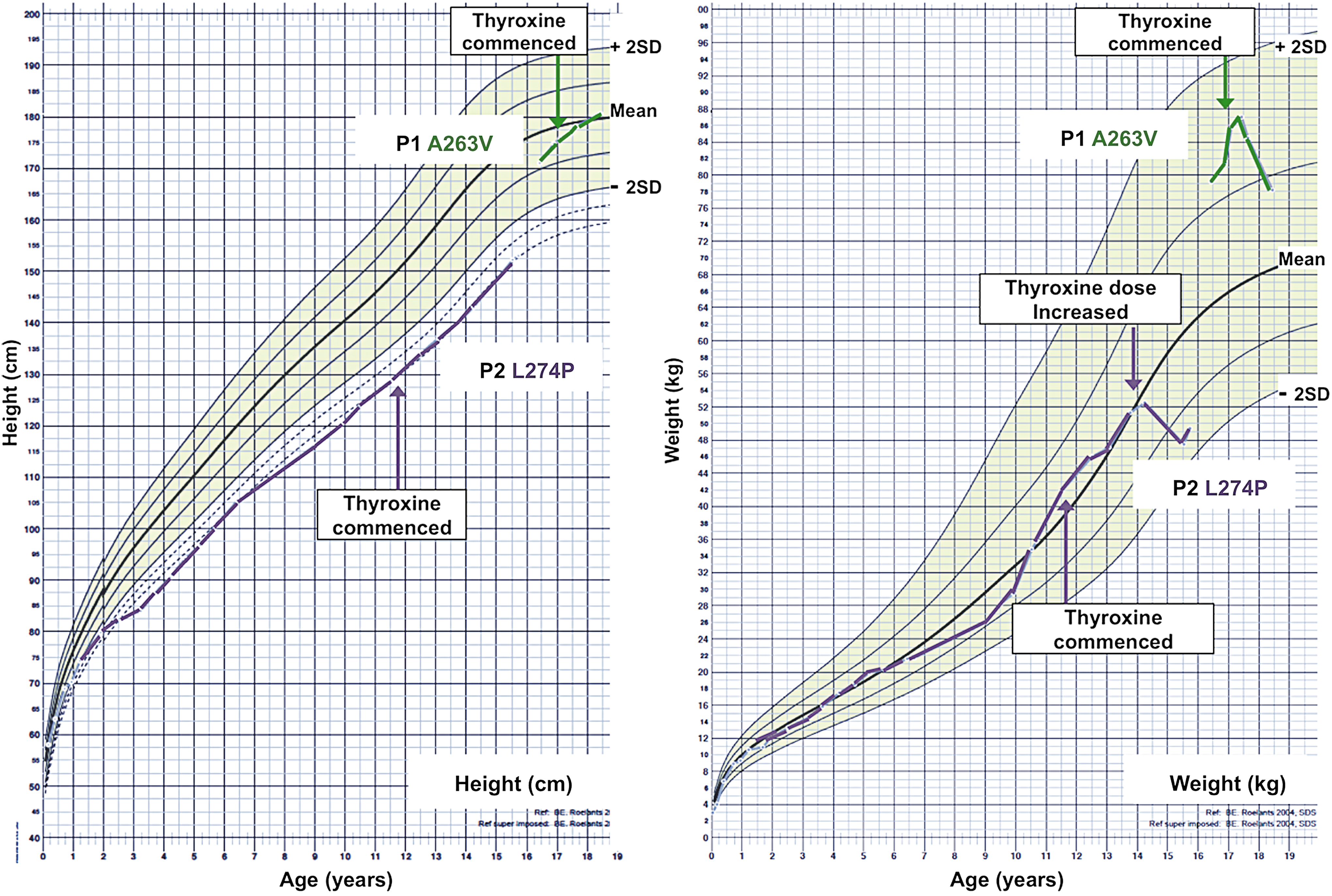
Charts showing height (left panel) or weight (right panel) standard deviation scores (normal range shaded), with serial measurements in patients P1 (A263V mutant TRα) or P2 (L274P mutant TRα). Arrowed points denote commencement of thyroxine treatment.
Following T4 treatment, resting energy expenditure was either unchanged or reduced on one occasion in P1 or slightly increased in P2, and sleeping heart rate was unchanged in both patients. Muscle CK and serum cholesterol (total and low-density lipoprotein) levels fell, with a slight rise in serum sex hormone binding globulin in both cases. Subnormal hematocrit in both patients remained unchanged (Table 2).
After T4 therapy, patient P1 reported marked changes, including better concentration and school performance, greater motivation and social confidence, improved coordination, and decreased constipation. Although patient P2 also reported greater regularity of bowel habits on T4 therapy, his alertness, energy levels, cool peripheries, and abnormal gait were unchanged.
Discussion
Two adolescent patients (P1 and P2) with THRA mutations were studied, and it was found that in each case, their clinical features and response to T4 therapy correlate with the functional properties of underlying mutant TRα.
P1 (A263V mutant TRα), relatively asymptomatic and diagnosed when investigated for possible delayed puberty, was of proportionate stature, slightly dysmorphic, and with mild neurocognitive impairment. In contrast, P2 (L274P mutant TRα) was investigated extensively in childhood for problems including neurodevelopmental delay and skeletal dysplasia and is growth retarded, quite dysmorphic, and markedly dyspraxic with cognitive deficit. Other features (macrocephaly, delayed dentition and bone age, constipation, and anemia) are common to both cases (Table 1).
In functional studies, A263V mutant TRα1 is transcriptionally impaired and inhibits function of its WT counterpart at low T3 levels (0.01–10 nM), but higher T3 concentrations (>100 nM) reverse such dysfunction and dominant negative inhibition, both in cotransfection assays in vitro and mutation-containing, patient-derived, PBMCs studied ex vivo. In contrast, only very high T3 concentrations (10 μM) reverse dysfunction of L274P mutant TRα1 in vitro, with failure to reverse subnormal KLF9 expression in patient's cells studied ex vivo. Structural modeling provides an explanation for such divergence in properties. The steric clash created by mutating alanine 263 to valine could be accommodated by slight repositioning of ligand (T3) (Fig. 6). However, the effect of mutating leucine 274 to proline would be much more dramatic because in the WT TRα1, the non-polar side chain of leucine 274 forms a hydrophobic buttress with the side chains of isoleucine 299 and phenylalanine 300 in helix 8, enabling the delta methyl group of isoleucine 299 to interact with T3 ligand. Substitution of proline for leucine 274 would no longer buttress isoleucine 299 weakening its interaction with T3, clash sterically with the sidechain of phenylalanine 300, perturbing the position of helix 8, and disrupt hydrogen bonding within the beta sheet in which it is located, perturbing normal interaction of leucine 276 and serine 277 with ligand (Fig. 6).
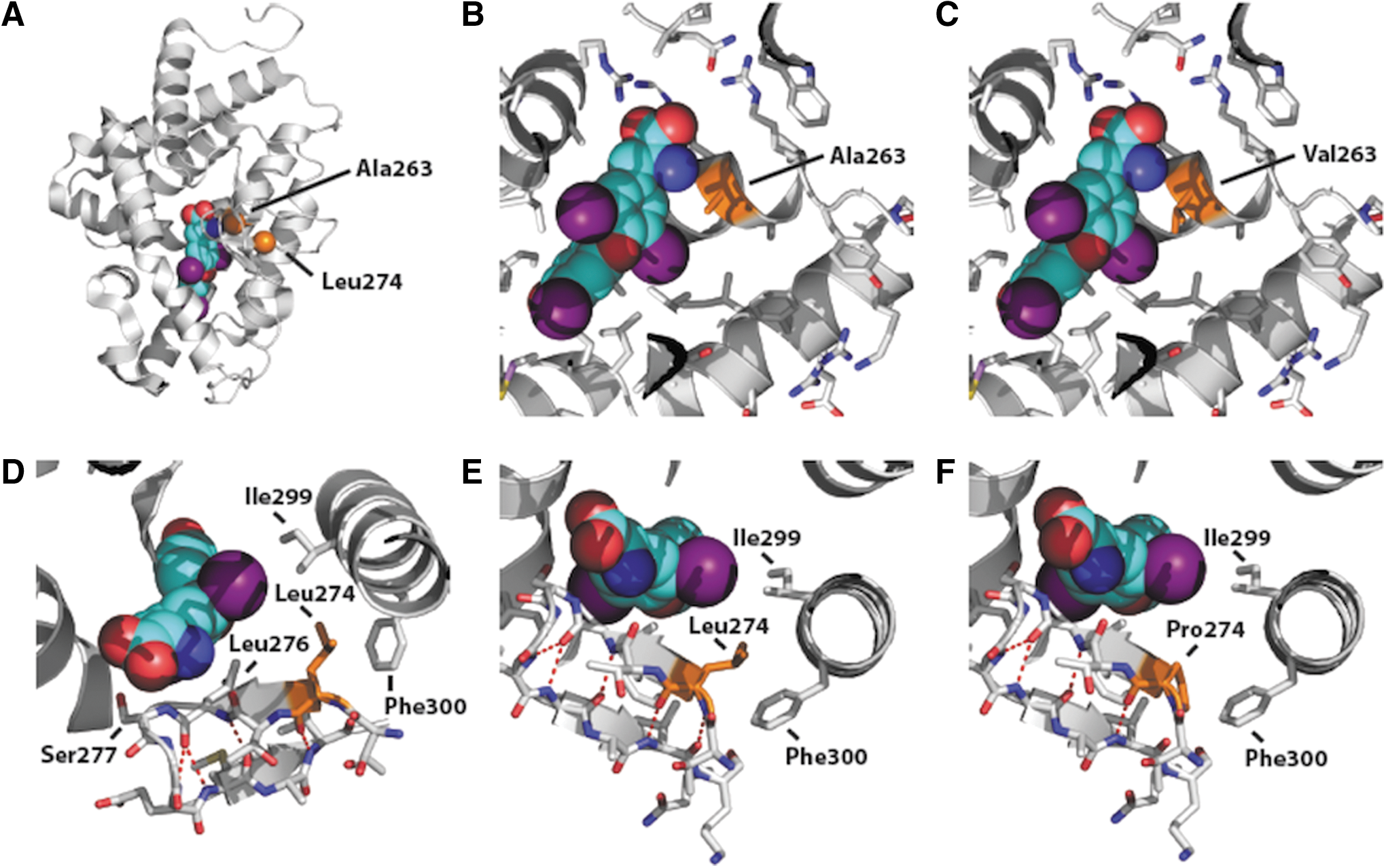
Crystal structure of the ligand binding domain of TRα1. The backbone of ligand (T3) is shown in blue with atoms in different colors (purple: iodine; red: oxygen); selected amino acid side chains are shown, colored by atom type; the location and sidechains of residues (Ala 263, Leu274) and the amino acids they are mutated to are shown in orange. Hydrogen bonds are indicated by dashed red lines (PDBID 2H77). (
Differences in T3-dependent reversibility of mutant receptor dysfunction in vitro are also correlated with differential responses of patients to T4 therapy: P1 (A263V mutant TRα) showed a change in linear growth, striking reduction in weight (fat mass), and improved dyspraxia, educational performance, and social engagement, whereas linear growth, motor incoordination, and mental alertness were unchanged in P2 (L274P mutant TRα), with reduction in weight only after increase in T4 dose.
The receptor defects identified in these patients increase the repertoire of RTHα-associated TRα mutations described hitherto (10), enabling relationships between genotype and phenotype to be further discerned. A mild phenotype in three individuals with A263V mutant TRα was previously documented (6), but they had been treated with T4 since very early childhood. However, mild clinical features in P1 who was not treated with T4 until late adolescence and in seven members of another family with a different amino acid change (A263S) at this locus (9) suggest that the phenotype associated with this mutant TRα genotype is intrinsically mild. It is also noteworthy that both receptor mutations reported here occurred de novo, such that 9 of the 14 TRα mutations recorded hitherto have occurred sporadically. As has been documented in RTHβ (11), this raises the possibility that more frequent occurrence of changes in mutation-prone nucleotides will result in certain codon changes being overrepresented in this disorder.
The L274P TRα mutation in P2 has a counterpart, with an amino acid change in the equivalent TRβ residue (L328S) in RTHβ (12). Indeed, most RTHα-associated TRα mutations described to date have an RTHβ-associated TRβ counterpart (10), raising the possibility that RTHα-associated TRα mutations will localize to particular regions within the receptor protein, as has been documented in RTHβ.
Although both the TRα mutations identified in the present patients also involve the TRα2 isoform, no added phenotype attributable to this could be discerned, and no gain or loss of function could be documented with the introduction of these amino acid changes into the TRα2 background. Thus, with the exception of a single case in which unusual skeletal and other features were recorded (7), no readily recognizable additional phenotypes could be identified in 14 individuals harboring mutations common to both TRα1 and TRα2 proteins (6,8,9). However, the possibility cannot be discounted of a mutation in TRα2 being associated with a phenotype that is either so subtle or of variable penetrance that it can only be discerned in a larger cohort of RTHα cases.
T4 therapy was permissive for increase in linear growth in P1 and also had beneficial metabolic effects in both him and three other individuals with the same TRα mutation (6): marked improvement in hypotonia, motor skills, and growth was recorded following T4 treatment at 18 months in an infant with another T3-reversible TRα mutation (D211G) (8). In a murine model of RTHα, harboring a missense (R384C) TRα1 mutation with T3-reversible dysfunction, raising circulating TH levels even in adult life, alleviated locomotor and behavioral abnormalities (13). Interestingly, as in patient P2, constipation has been noted to improve, even in cases with a highly deleterious TRα defect (3,4). Overall, these observations suggest that a trial of T4 therapy is warranted in RTHα with mild mutations, as has been advocated by others (14), and that it could alleviate some symptoms, even in patients with a severe receptor defect. However, as observed in P1 and P2, suppression of TSH, fall in serum cholesterol, and rise in sex hormone binding globulin levels after T4 therapy (4,6) do suggest thyromimetic effects in TRβ-expressing tissues (e.g., pituitary, liver), and the long-term consequences of this are unknown. Perhaps this provides the rationale for development of TRα-selective analogues that are potentially devoid of such effects.
Footnotes
Acknowledgments
Our research is supported by the Wellcome Trust (Investigator Award 095564/Z/11/Z to K.C.; Investigator Award 100237/Z/12/Z to J.S.) and NIHR Cambridge Biomedical Research Centre (C.M. and K.C.). J.S. is a Royal Society Wolfson Research Merit Award Holder.
Author Disclosure Statement
The authors have nothing to declare.