
Abstract
Background:
Euthyroid multinodular goiter (MNG) is common, but little is known about the genetic variations conferring predisposition. Previously, a family with MNG of adolescent onset was reported in which some family members developed papillary thyroid carcinomas (PTC).
Methods:
Genome-wide linkage analysis and next-generation sequencing were conducted to identify genetic variants that may confer disease predisposition. A multipoint nonparametric LOD score of 3.01 was obtained, covering 19 cM on chromosome 20p. Haplotype analysis reduced the region of interest to 10 cM.
Results:
Analysis of copy number variation identified an intronic InDel (∼1000 bp) in the PLCB1 gene in all eight affected family members and carriers (an unaffected person who has inherited the genetic trait). This InDel is present in approximately 1% of “healthy” Caucasians. Next-generation sequencing of the region identified no additional disease-associated variant, suggesting a possible role of the InDel. Since PLCB1 contributes to thyrocyte growth regulation, the InDel was investigated in relevant Caucasian cohorts. It was detected in 0/70 PTC but 4/81 unrelated subjects with MNG (three females; age at thyroidectomy 27–59 years; no family history of MNG/PTC). The InDel frequency is significantly higher in MNG subjects compared to controls (χ2 = 5.076; p = 0.024. PLCB1 transcript levels were significantly higher in thyroids with the InDel than without (p < 0.02).
Conclusions:
The intronic PLCB1 InDel is the first variant found in familial multiple papilloid adenomata-type MNG and in a subset of patients with sporadic MNG. It may function through overexpression, and increased PLC activity has been reported in thyroid neoplasms. The potential role of the deletion as a biomarker to identify MNG patients more likely to progress to PTC merits exploration.
Introduction
E
Familial non-medullary thyroid cancers account for about 5% of thyroid cancers and have a younger age of onset than sporadic disease. They are associated with four susceptibility loci (13 –16) on chromosomes 19p13.2, 2q21, 1q21, and 10q23 (PTEN). There is some overlap with familial goiter in which eight predisposing loci have been identified (12,17 –20) on chromosomes Xp22, 3q26, 2q, 3p, 7q, 8p 14q13.3, and 14q32, the last two including the NKX2.1 (21) and the RNAse DICER1 genes, respectively (22). A role for the predisposing loci on chromosomes 2q.35, 5q.24, 8p.12, and 14q.13 has been confirmed in Chinese families (23). Genes implicated in familial goiter and cancer generally differ from those in sporadic disease, with the exception of NKX2.1 (21) and FOXE1 (24).
Previously, a family exhibiting a type of euthyroid MNG was reported (25), with papillary adenomas of adolescent onset affecting eight individuals in four generations to date. MNG is known to have progressed to PTC in two of the eight affected family members. Microsatellite analysis was applied to exclude loci described above on chromosomes 14q, Xp, 3q, 9p, 2q, and 1q. Since one family member had co-existing breast cancer and another co-existing kidney disease, genes co-expressed in these tissues and the thyroid—NIS and PAX8, respectively—were investigated. Sanger sequencing revealed no abnormality in either gene. Subsequently, the PTEN gene has been fully sequenced in the family member with breast cancer, and no mutations were detected.
The aim of this study was to apply genome-wide linkage analysis (GWLA) and next-generation sequencing (NGS) to identify the gene variant(s) responsible for the observed phenotype in this family. The study then aimed to assess the frequency of any variant(s) detected in other relevant cohorts.
Methods
GWLA
A GWLA of the family described in (25) and summarized in Figure 1 was undertaken. All patient samples were obtained with informed consent and Local Research Ethics Committee (LREC) approval. Genomic DNA was extracted from the whole blood of 18 family members (those labeled in the tree), of whom eight were affected (seven females), according to the manufacturer's instruction (Qiagen) and quantified using a Nanodrop. Samples (250 ng) were processed following the manufacturer's protocol and the DNA integrity monitored by agarose gel electrophoresis before being hybridized at 48°C for 18 hours to Affymetrix GeneChip™ Human Mapping 10K 2.0 Arrays. The chips were scanned using an Affymetrix GeneChip scanner 3000. Data were acquired using GeneChip® Operating Software and analyzed using GTYPE software.

Family tree. The index patient (IV 6) was referred in 2002 aged 12 to the University Hospital of Wales (Cardiff, United Kingdom) with multinodular goiter (MNG). She had a normal 46 XX karyotype; thyroid function and autoantibody tests were all within reference ranges, confirming euthyroidism. A thyroid ultrasound (US) scan showed multiple hyperechoic nodules. Fine-needle aspirate (FNA) histology revealed multiple papilloid adenomata lacking features of papillary carcinoma. She underwent total thyroidectomy, and her kidney scan was normal. A thyroid US of her 18-year-old brother (IV 3) revealed hyperechoic cysts, and FNA showed changes suggesting papillary thyroid cancer (PTC). He underwent total thyroidectomy, and the microscopic examination showed follicular nodules (with complex folding of thyroid epithelium in some nodules) but no true PTC. In infancy, he had a nephrectomy for polycystic kidney disease. The other five siblings also underwent a thyroid US and kidney scan. The index patient's sister (IV 1) had MNG, but a thyroid scan revealed small hypo-echoic cysts. She underwent thyroidectomy in 2016, but her son (shown in the tree as generation V, but not labeled) developed MNG in 2014 at the age of 5 years (not genotyped). The index patient's younger sister (IV 7) was pre-pubertal in 2002 but has since developed MNG at the age of 18 years while (IV 4) developed Graves' disease. Both underwent thyroidectomy. The brothers (IV 2 and IV 5) had no thyroid or kidney lesions aged 24 and 13, respectively. The mother of the index patient (III 2) underwent partial thyroidectomies at the age of 17 and 27 for MNG and a total thyroidectomy at 28 when PTC was present. She developed breast carcinoma at the age of 41 years and died of this in 2013 aged 52. The maternal uncle (III 3) and his family (III 4 and IV 8) have no thyroid or kidney lesions, but the maternal grandmother (II 2), great-grandmother (I 2), and mother's first cousin (III 5) underwent surgery for presumed benign thyroid disease. Individuals with solid symbols had MNG, and all labeled family members (n = 18) were genotyped. The InDel is present in all individuals with MNG (with solid symbols) and also II 3, IV 2, and IV4. Only the mother (III 2) was confirmed to have PTC. Underlined numbers indicate individuals carrying the InDel.
Two quality-control steps were performed. The first eliminated single nucleotide polymorphisms (SNPs) showing “no call” in more than four individuals. The second step would have eliminated data from any individual with >10% “no calls,” but this did not apply, and the data of all 18 family members were retained. Graphical representation of relationships software was used to determine how many alleles are shared (identity by state) at each locus. Mendelian errors were tested using PedCheck software. PLINK was used to merge family data (founders) with HapMap to investigate ethnicity. Multidimensional scaling was performed on the family merged with HapMap data from 60 European individuals (CEU), 90 Chinese and Japanese, and 60 Yoruba. The family was closest to the European cluster (data not shown). Thus, allele frequencies were based on CEU HapMap data. Using MERLIN software, the primary analysis was multipoint nonparametric and the secondary analysis multipoint parametric dominant mode, assuming 90% penetrance in females, 50% in males, and age of onset later than 12 years (based on clinical information summarized in Fig. 1). Single-point analyses were also used to support the findings of multipoint analysis. Since data are derived from a single large family, there is considerable allele sharing, and hence the Kong and Cox exponential (–exp) model was used (for nonparametric analysis) (26).
Haplotype analysis
MERLIN software (–best) was also used to perform a haplotype analysis in the region of maximum LOD score on chromosome 20. The haplotype was also confirmed manually.
Copy number variation analysis
Genomic DNA for copy number variation (CNV) analysis of the index patient was quantified and prepared for hybridization to Illumina Human 660W-Quad BeadChips according to the manufacturer's instructions. Data were analyzed using PennCNV (27) software. CNVs were required to be 1 kb long and cover at least 10 consecutive markers (SNP or cnvi) to be considered positive. The study focused on the region with a high LOD score identified in the GWLA.
NGS
Primer pools for preparation of DNA libraries were designed using Ampliseq 3.0.1 software (
NGS was performed on 98 individuals: all 18 family members plus 80 unrelated subjects with MNG (please see below).
Other variants identified in the family using NGS were interrogated in the Study of Health in Pomerania (SHIP) cohort (28). Relevant genotyping data were available from 986 individuals who were either unaffected or presented with diffuse goiter (as defined in Teumer et al.) (29) and/or MNG (nodules identified by ultrasound). Figure 2 details the filtering steps and evaluations undertaken to assess whether detected variants might be linked with disease.
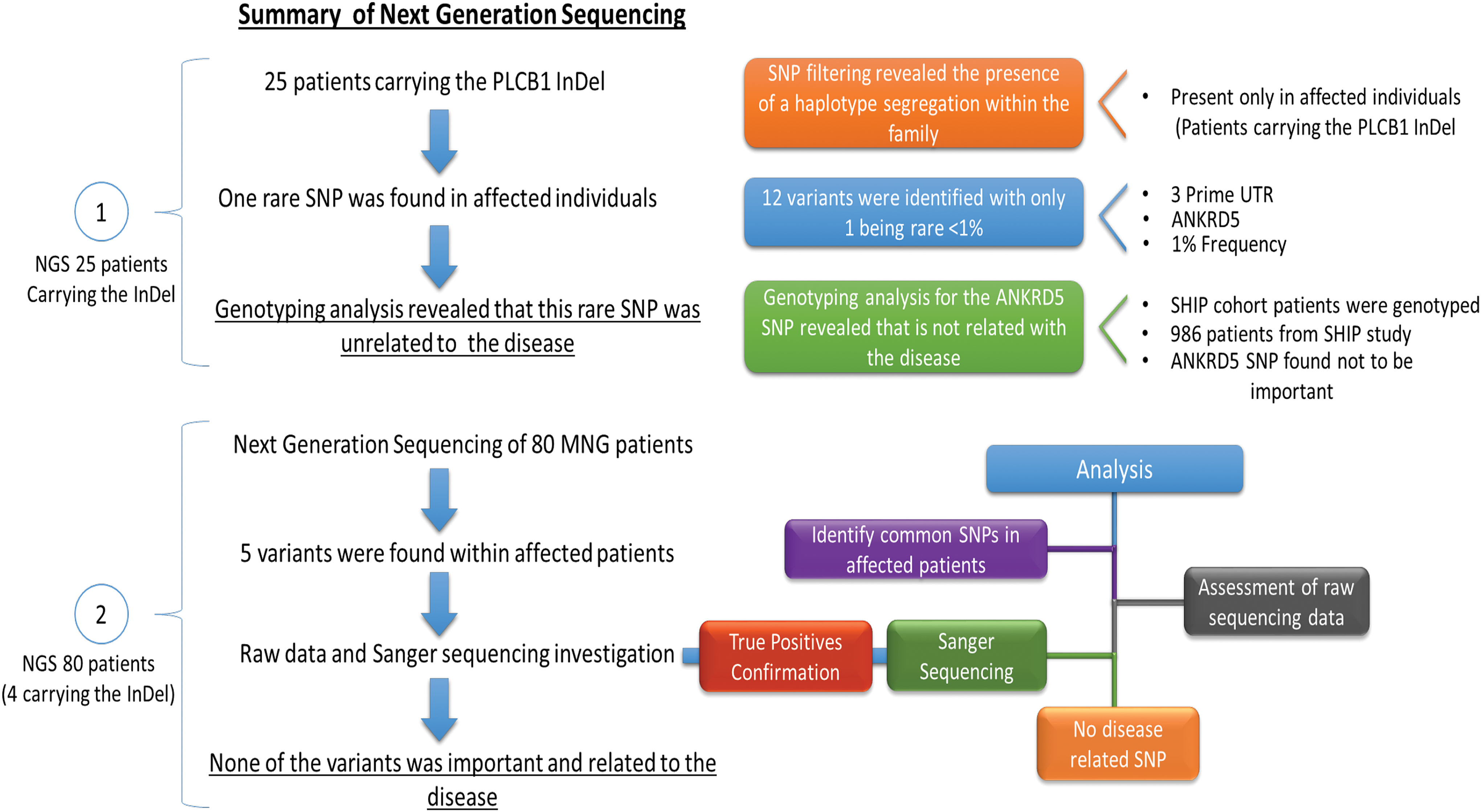
Filtering steps conducted to evaluate variants identified by next-generation sequencing. Color images available online at
Defining deletion frequency
Primers within and flanking the deleted region were designed using Primer 3 software (Supplementary Table S1b) for PCR amplification of genomic DNA from all family members and 105 unrelated euthyroid individuals from the United Kingdom. PCR amplicons were analyzed by agarose gel electrophoresis and polyethylene glycol precipitated for Sanger sequencing using Big Dye Terminator Cycle Sequencing Ready Reaction (ABI Prism; PE Biosystems) and analysis on an ABI 3100 Genetic Analyzer.
Tissues from patients recruited in Australia (snap frozen and stored in liquid nitrogen) were also studied and consisted of 70 PTC and 81 MNG patients (ethics approval from the Northern Sydney Area Health Service Human Research Ethics Committee). To avoid population stratification, only subjects with self-reported white European ancestry were included. Patient data and tissues were collected between 1992 and 2012 at the Kolling Institute of Medical Research. Genomic DNA for genotyping was obtained from thyroid tissue using Qiagen kits and analyzed by PCR and Sanger sequencing as described above. These samples also underwent NGS.
High-throughput screening of PLCB1 InDel, analysis of additional cohorts
A qPCR-based genotyping tool was developed using primers within and flanking the PLCB1 InDel, as described above (Supplementary Table S1b). The genotyping tool was used to screen 200 breast cancer patients. Initial optimization experiments revealed that greatest specificity was obtained using primers flanking the InDel. The qPCR obtained a difference of approximately 10 Ct for samples with and without the InDel. The qPCR was performed with approximately 100 ng of genomic DNA Input, 1 × SyBR green master qPCR mix (Invitrogen), and 100 nM of each primer in a 25 μL reaction. qPCR conditions included an initial hold step at 50°C for 2 min, then 95°C for 2 min followed by 40 cycles of 95°C for 15 s and 60°C for 30 s, then a hold step at 95°C for 1 min, 55°C for 30 s, and 95°C for 30 min. Samples found to harbor the InDel by qPCR were confirmed by Sanger sequencing.
Transcript measurements of PLCB1 isoforms
Thyroid tissue was obtained from three affected family members heterozygous for the InDel and five subjects undergoing thyroidectomy for autoimmune thyroid disease expressing two normal PLCB1 alleles (all confirmed by genotyping). Thyroid RNA was extracted and reverse transcribed using standard protocols, and qPCR (SYBR Green incorporation measured on a Stratagene MX 3000) was used to measure transcript levels and evaluate proportions of PLCB1-a and PLCB1-b isoforms (primers in Supplementary Table S1c; wild-type amplicon identity confirmed by Sanger sequencing). Comparison with standard curves for transcript levels of isoform 1a and 1b permitted calculations of absolute values for each sample. Transcripts for a housekeeping gene (APRT) were also measured, and values were expressed relative to this (transcripts/1000 APRT). In a single qPCR experiment, all measurements were made in duplicate; the standard curve was also run in each reaction. Transcript levels of the various PLCB1 isoforms were compared between deletion affected and non-affected thyroids using the Mann–Whitney U-test, and differences where p < 0.05 were taken to be significant.
Results
Genome-wide linkage, haplotype, and CNV analyses
A multipoint nonparametric LOD score of 3.01 was obtained over 19.5 cM on chromosome 20p (Fig. 3 and Supplementary Fig. S1). In secondary analysis, the same region gave a multipoint dominant LOD score of 2.16, based on a disease model with 0.01 allele frequency, 50% penetrance for males and 90% for females, both aged >12 years. LOD scores on the remaining 21 autosomes and X chromosome were all <1 (Fig. 3). Single-point analyses supported the multipoint data for both nonparametric and model-based linkage on all chromosomes (Supplementary Table S2).
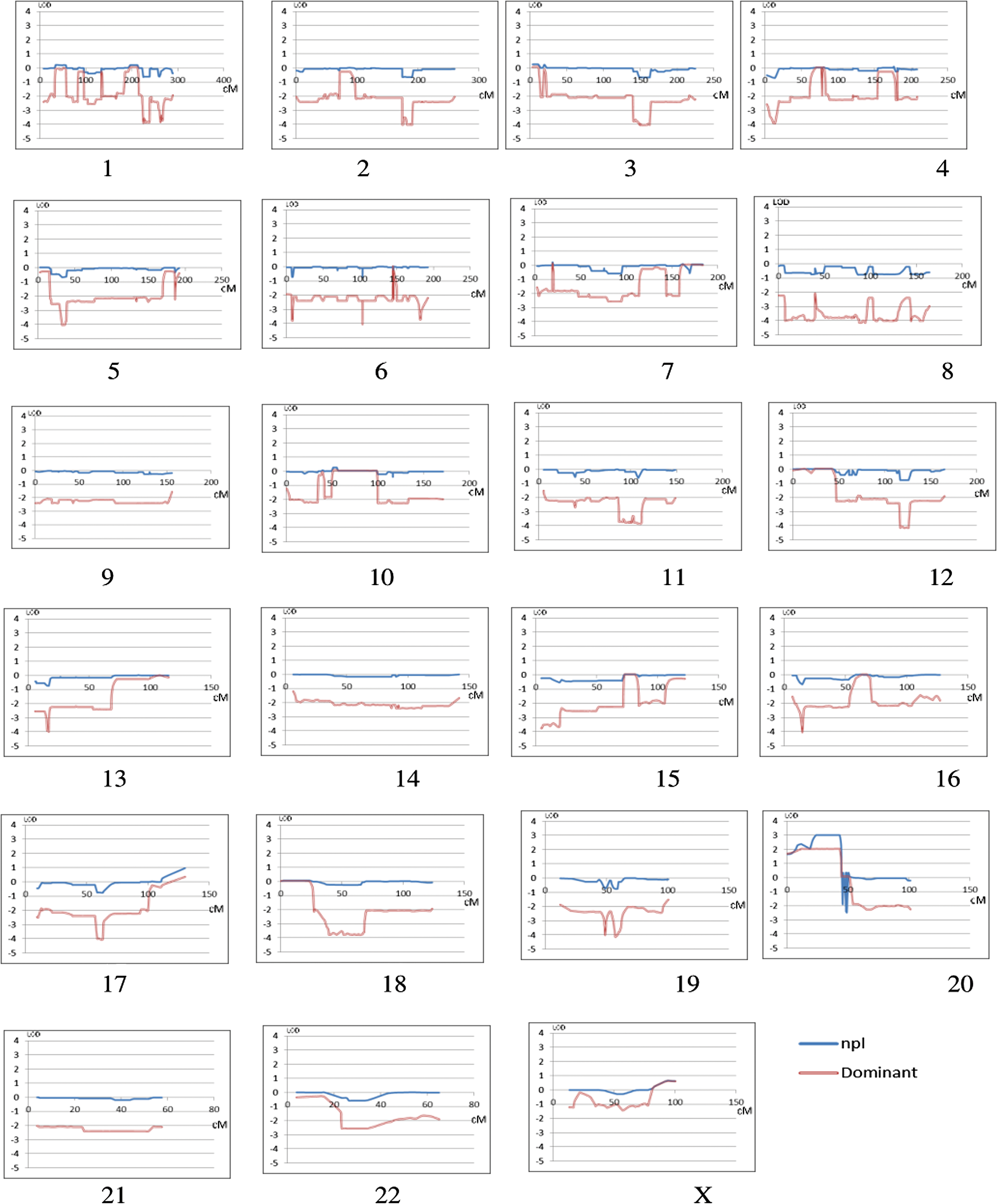
LOD scores of all chromosomes. Plots of all chromosomes showing the maximum LOD scores obtained (y-axis) in multipoint nonparametric (blue line) and multipoint dominant analyses (red line) based on a disease model with 0.01 allele frequency and 50% penetrance for males and 90% for females, both age >12 years. The x-axis shows the position on each chromosome in cM. In all cases, the single-point analysis supported the results of the multipoint. Color images available online at
Haplotype analysis was employed to identify a possible disease locus and reduced the region of interest to 8.73 cM (3.7 Mbp), which includes 10 genes (Fig. 4, Supplementary Figs. S2 and S3). The haplotype was not found in 503 individuals from the 1000 Genomes European data set, although one individual missed only the last marker suggesting a shorter version of the haplotype (red highlight in Supplementary Fig. S3A).

Haplotype analysis of defined region. Haplotypes estimated by MERLIN software using (–best command) to obtain haplotypes corresponding to the most likely gene flow. MERLIN labeled founder's haplotypes alphabetically and then listed the two haplotypes for each individual, maternal first, paternal second. Color images available online at
Analysis of CNV in an affected individual revealed a deletion of approximately 900 bp located in the third intron in one copy of PLCB1 in the region of interest (Supplementary Fig. S4; the log R ratio mean was −0.451, over 14 markers, with at least one marker below −1.00).
Defining the deletion frequency in the family and selected cohorts
The length of the deletion was confirmed to be 1077 bp by standard PCR and Sanger sequencing, using primers flanking and within the deletion, to reveal one copy of full-length and one deleted allele in all affected and obligate carrier II-3 but only the full-length product in family members free of any sign of MNG. The sequence of the allele bearing the deletion corresponds to that immediately upstream and downstream of the deleted region but with an additional “ATAA” inserted at the junction. Hence, it is an InDel.
Standard PCR was applied to genotype a selected cohort of 105 Caucasians in whom thyroid function testing was clinically indicated because of general fatigue. A woman in her forties, with no history of thyroid disease, was heterozygous for the InDel. Further in silico analyses using the database for genomic variants (30) identified a report that detected the InDel (variation 67651, LRR −0.645) in 2/180 Caucasians but none in >450 people of other ethnicities (31). Combining the present genotyping data with that of Conrad et al. (31) reveals 3/285 Caucasians harboring the InDel, suggesting that it is relatively rare (around 1%).
Subsequently, genomic DNA was extracted from thyroid tissue from 70 patients undergoing surgery for non-familial PTC and an additional 81 operated for non-familial MNG. PCR analysis was used to test for the InDel, as described above. The InDel was not detected in any of the PTC patients, but 4/81 MNG were heterozygous for the InDel, and sequencing revealed the same ATAA insertion at the junction. Comparison of the frequency of the InDel in the general population with that in MNG gives a chi-square value of 5.076 (one degree of freedom; p = 0.024, two-tailed). The four MNG patients (three female) are unrelated and with no apparent family history of MNG or PTC at the time of their surgery. The age at thyroidectomy was between 27 and 59 years, and the pathology is variously described as “oncocytic neoplasm with variable patterns of growth” to “cystic degeneration with calcification.” The study also investigated whether the PLCB1 InDel might be implicated in breast cancer using the qPCR-based screening protocol. Prevalence in this cohort was similar to that of the general population (i.e., 1%), since just two breast cancer patients harbored the PLCB1 InDel.
NGS of the Chr20 high LOD score region
The proton sequencer generated 9.9 Gbp of data, achieving 98% accurately mapped sequences with >88% of the percentage of target bases covered by at least 0.2 times the average base read depth.
A total of 181 sequence variants between Chr20 8113405 and 11907285 were identified in the family, with the minor allele being on the disease risk haplotype in 12 of these. Given the rarity of PTC and the expected high penetrance, a pathogenic variant is expected to have a very low population frequency. After referring to the UCSC genome browser, only 1/12 variants was found to have a minor allele frequency <1%. Its presence in affected family members was confirmed by Sanger sequencing. The variant is at Chr20 10036484 (rs56234782), with T (98.8%) or C (1.2%) in the 3′ UTR of the ANKRD5 gene. To investigate whether it is implicated in goiter and/or thyroid nodule formation, its frequency was investigated in the SHIP cohort. However, even though the minor allele was more prevalent in the entire cohort, the prevalence in the affected population (goiters 1.9% and nodules 2.54%) was lower than in the unaffected populations (2.79% and 2.85%, respectively), thereby excluding a role for it in MNG.
The MNG cohort was also submitted to NGS analysis. This identified >300 different sequence variants across the 80 patients. However, all were also present in the 1000 Genomes cohort at a population frequency of >1%. It was therefore considered unlikely that any of these variants are pathologically relevant to MNG, thereby confirming the relevance of the InDel.
Transcript measurements of PLCB1 and effect of knockdown on thyroid growth
Having confirmed that the InDel may contribute to the pathogenesis for MNG (perhaps in combination with other factors), the study investigated how it might promote thyrocyte proliferation. The InDel is in the large third intron of PLCB1, the phosphoinositide-specific enzyme that generates IP3 and DAG, leading to PKC activation, and also links signaling between the MAPK cascade and G protein coupled receptors (32). PLCB1 is present in several isoforms, including PLCB1-a and PLCB1-b, with the latter having a predominantly nuclear location (33). To test the hypothesis that the InDel causes preferential transcription of certain PLCB1 isoforms, RNA was extracted from thyroids from the original family and from subjects undergoing thyroidectomy for benign disease. In all cases, genomic DNA from the donor thyroid was tested for the PLCB1 deletion.
qPCR analysis of InDel-affected thyroids did not indicate altered expression of the major PLCB1 isoforms a and b (sequenced to confirm they were wild type; data not shown). However, qPCR measurements indicated significantly higher PLCB1 transcript levels (p < 0.02) in thyroids from family members with the InDel compared to those from benign thyroid disease who do not harbor the variant (Fig. 5). Lack of thyroid tissue precluded analyzing PLCB1 protein levels.
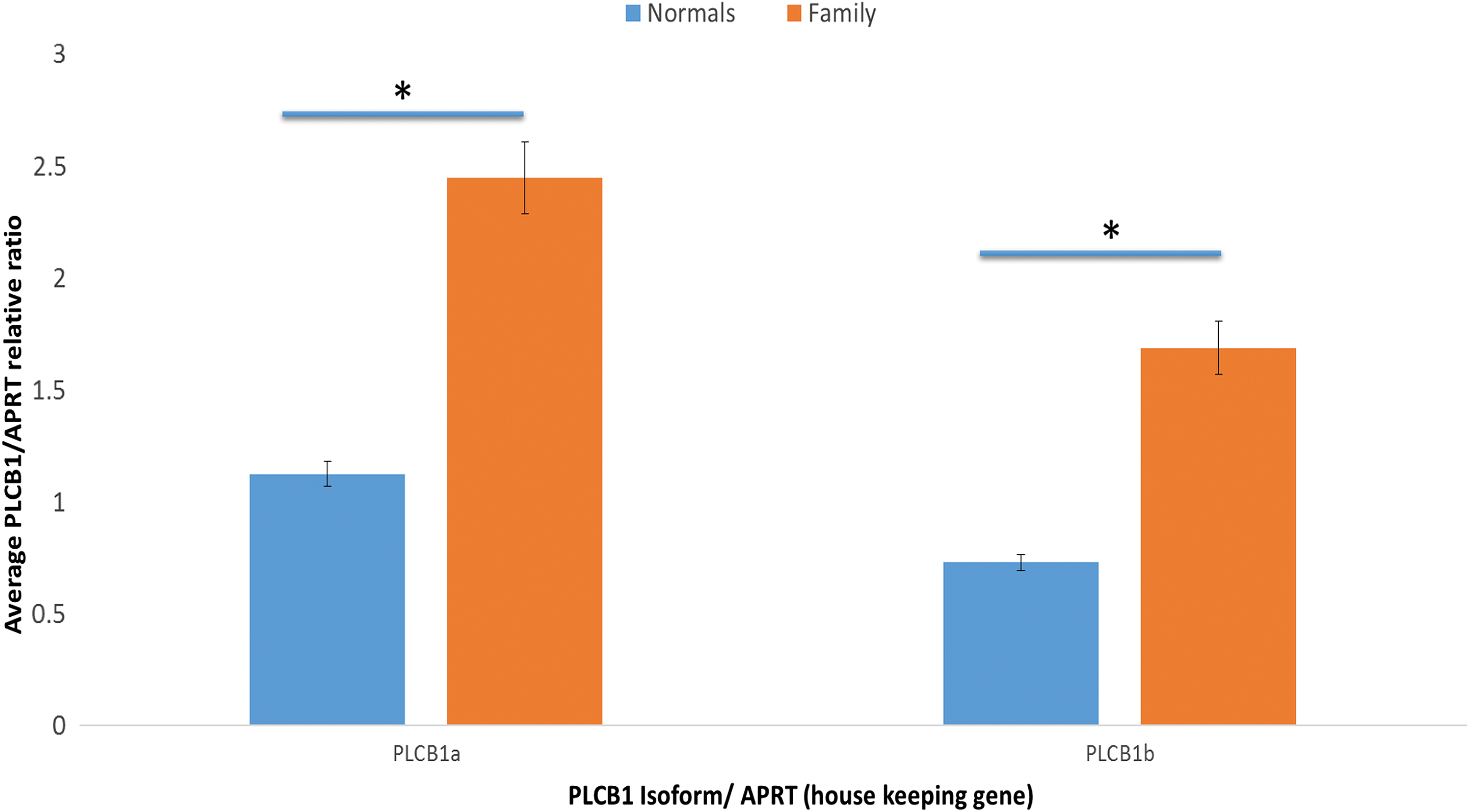
Quantitative polymerase chain reaction (qPCR) measurement of PLCB1 isoforms. Expression of PLCB1-a and PLCB1-b isoform transcripts in human thyroids from deletion-affected (n = 3) and non-affected (n = 5) individuals. RNA was extracted from thyroid tissue and reverse transcribed using standard protocols and transcripts for PLCB1-a and PLCB1-b measured by qPCR. Results are reported relative to the APRT housekeeping gene and are the mean ± standard deviation of three independent experiments in which all samples were measured in duplicate. Results indicate significantly higher transcript levels of isoform a in all thyroids tested (p < 0.01) but no significant difference in the ratio of PLCB1-a to PLCB1-b in deletion-affected and non-affected thyroids. However, transcript levels of both PLCB1-a and PLCB1-b are significantly higher (p < 0.02) in deletion-affected thyroid tissues. Color images available online at
Discussion
The GWLA led to the identification of an InDel in the family with a type of MNG, located in the large third intron of PLCB1, a gene encoding an enzyme with a central role in several signaling cascades involved in regulating thyrocyte growth. Subsequent NGS in the family failed to identify any other disease-linked variant, thus supporting a role for the PLCB1 InDel in the pathogenesis of MNG in this family.
The InDel comprises a loss of 1077 bp, with an ATAA inserted at the junction in all affected family members and the four unrelated patients with MNG. It is suggested that this may indicate a “cut and paste” event indicating transposon activity. Interestingly, a 11 kb transposon cluster has been identified immediately upstream of the 3.7 Mbp section on chr 20, displaying a nonparametric LOD score of 3.01 in the current study (34). Of note, the LOD score of 3.01, while at the lower limit to be considered significant, is higher than the maximum estimated for a kindred, having eight affected individuals (35).
The same InDel was detected in one subject of a selected cohort of 105 people in whom measuring thyroid function was clinically indicated. The database of genomic variants was also consulted, and several reports of relevance were found. Conrad et al. found the deletion in 2/180 Caucasians, but insufficient detail is provided to know whether it is a simple CNV or the same InDel identified in the present studies. Combining the genotyping data with that of Conrad et al. reveals that 3/285 Caucasians harbor the deletion, suggesting that it is rare (31). Several other authors did not observe this deletion, but aware of the difficulty in detecting small CNVs, these were not included in the current calculation. In addition, 200 patients with breast cancer have been screened for the InDel, with only two harboring this deletion. Hence, the prevalence was similar to the general population, suggesting that there is no connection of the InDel with breast cancer.
Next, the study considered how the deletion or novel PLCB1 InDel might exert its effects. The region was explored using the Encyclopedia of DNA Elements (although compiled without inclusion of thyroid tissue or cell lines) (36), which revealed the existence of a binding site for the estrogen receptor alpha (ERα) within the deletion. This is of potential importance, since all thyroid diseases are more prevalent in women than men (1). The incidence of thyroid disorders increases in the years immediately following puberty, and in vitro studies have demonstrated that estrogen can promote thyrocyte proliferation (37) by several mechanisms. The PLCB1 InDel is located in an intron. While many functional transcription factor binding sites are found in promoters, a systematic search for ERα binding sites in the human genome identified >1000, with >95% of them residing in introns and not promoters (38).
Experiments were also conducted to determine whether the deletion alters the ratio of PLCB1-a and PLCB1-b, which are generated by alternative splicing. Differences in their C terminal sequence mean that only PLCB1-a has a nuclear export signal. No alteration was found in the ratio of PLCB1-a and b isoforms, but in all cases, transcript levels for PLCB1 were higher in thyroids from people heterozygous for the InDel than in thyroids with two full-length copies. This suggests that the InDel may contribute to MNG development through overexpression of PLCB1. Furthermore, total PLC enzyme activity is elevated in thyroid neoplasms (39), but unfortunately PLC inhibitors lack the specificity required to identify which isoform is responsible. Increased PLCB1 expression has also been reported in small-cell lung carcinoma (40), and expression of PLCB2 is substantially increased in breast cancer and is used as a prognostic marker (40).
As mentioned above, PLC enzymes activate PKC, and genes implicated in this signal pathway are upregulated in euthyroid MNG (41). They also link signaling via Gq (which can also be activated via the thyrotropin receptor) to the MAPK cascade, and the thyroid disruption of this pathway, by thyrocyte-targeted Cre/Lox P knockdown of the Gqα subunit, produces mice that are resistant to goiter formation when fed a goitrogenic diet (42). However, when Western blots were performed with protein extracts of thyroid tissue from family members with the PLCB1 InDel, surprisingly it was observed that pMAPK levels were substantially lower than in thyroid tissue from patients with autoimmune thyroid disease or MNG without the PLCB1 InDel (Supplementary Fig. S5).
In conclusion, the PLCB1 InDel identified in this family with MNG also occurs in a proportion of sporadic MNG, and may provide a biomarker to identify MNG patients more likely to progress to PTC. The PLCB1 InDel appears to predispose to goiter formation, possibly by increasing PLCB1 transcription with subsequent downstream effects.
Footnotes
Acknowledgments
We express our sincere gratitude to the members of the family who participated in this research. The work was part funded by the Government of Saudi Arabia (ref A390), by the Medical Research Council, and by the Onassis Foundation. SHIP is part of the Community Medicine Research Network of the University Medicine Greifswald, Germany (
The March 2006 human reference sequence (NCBI Build 36.1) produced by the International Human Genome Sequencing Consortium was used as a reference genome (UCSC Genome Browser;
Author Disclosure Statement
There is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.