
Abstract
Background:
Resistance to thyroid hormone due to THRA mutations (RTHα) is a recently discovered genetic disease, displaying important variability in its clinical presentation. The mutations alter the function of TRα1, one of the two nuclear receptors for thyroid hormone.
Methods:
The aim of this study was to understand the relationship between specific THRA mutations and phenotype. CRISPR/Cas9 genome editing was used to generate five new mouse models of RTHα, with frameshift or missense mutations.
Results:
Like human patients, mutant mice displayed a hypothyroid-like phenotype, with altered development. Phenotype severity varied between the different mouse models, mainly depending on the ability of the mutant receptor to interact with transcription corepressor in the presence of thyroid hormone.
Conclusion:
The present mutant mice represent highly relevant models for the human genetic disease which will be useful for future investigations.
Introduction
R
Understanding of RTHα greatly benefits from the knowledge of TRα1 structure and function (11). This nuclear receptor is a transcription factor composed of an N-terminal domain, which binds DNA, and a C-terminal domain, which binds T3. Unliganded TRα1 binds DNA and recruits protein complexes containing corepressors at the chromatin. Upon ligand binding, the short C-terminal α helix (helix 12) of TRα1 repositions, favoring the recruitment of coactivator complexes at the expense of corepressors. This results in a sharp transition from transcriptional repression to activation for numerous genes. All known THRA human mutations are in the C-terminal domain and fall into two categories. In the first category, amino acid substitutions (D211G, A263V, A263S, N359Y, H361Q, R384C, and R384H) reduce the affinity of TRα1 for T3. In the second category, C-terminal substitutions (P398R and E403K), truncations (C392X and E403X), and frameshift mutations (C380fs387X, A382PfsX7, F397fs406X, and F401S) not only alter ligand binding but also prevent coactivator recruitment. The autosomal dominant inheritance of RTHα is explained by the fact that all mutated receptors exert in vitro a dominant-negative activity toward intact TRα1 and probably TRβ1 as well. The molecular basis for this dominant-negative property is, however, unclear. It seems that the complexes repressing transcription, containing unliganded or mutated TRα1 and the NcoR corepressor, compete with coactivator-associated TRα1 for chromatin occupancy, leaving long-lasting repressive marks on histone tails (12).
Mouse Thra and human THRA genes display extensive sequence similarities. Alternate promoter usage, splicing, and codon usage produce non-receptor proteins in both species (13). The two TRα1 amino acid sequences only differ at three positions (AA34, 37, and 170). This makes mouse lines with Thra mutations highly relevant animal models for RTHα. Several years before the discovery of RTHα in humans, four knock-in alleles were built, introducing missense mutations in Thra (14 –17). In all but one case, Thra was extensively modified, and foreign sequences were inserted for commodity. Alternate splicing was altered, eliminating the non-receptor TRα2 protein, which does not bind T3, while mutating the TRα1 reading frame. Although these knock-in mutations are seemingly equivalent, the phenotypes are, for unknown reasons, very different and at odds with the human RTHα presentation (13). Reports of obesity (17), decreased fertility (14), extreme anxiety-like behavior (18), and high mortality (14,16) contrast with patient reports and raise questions about the relevance of these mouse models.
In order to investigate precisely the genetic basis of the phenotype variability of RTHα, both in humans and in mice, CRISPR/Cas9 genome editing was used to introduce five novel germline mutations (four frameshift and the N359Y missense mutation) in the mouse Thra gene, closely modeling the mutations found in RTHα patients. This approach avoided the introduction of superfluous exogenous sequences and allowed some of the mutations found in patients to be modeled. Phenotype analyses showed that these mutations have milder consequences than previous knock-in mutations, and that the new mouse strains are faithful models of the human diseases. It was also found that similar mutations, altering the same C-terminal helix of the receptor, have very different consequences, and a plausible molecular explanation for the phenotype variability was identified. Overall, these data greatly clarify the genotype/phenotype relationship in RTHα and confirm that a simple in vitro characterization can predict disease severity.
Methods
All oligonucleotide DNA and RNA sequences are listed in the Supplementary Data (Supplementary Data are available online at
Animals and gene editing
The research project was approved by a local ethic committee and subsequently authorized by the French Ministry of Research (license #6711), and carried out in accordance with the European Community Council Directives of September 22, 2010 (2010/63/EU) regarding the protection of animals. Generation of frameshift mutations was the result of the error-prone non-homologous end joining pathway, which repairs DNA after Cas9 cleavage. A 20-mer DNA fragment was inserted between the BbsI restriction sites of pX330 (Addgene plasmid #42230), as described (19). This generated an expression vector for both Cas9 and a single-guide RNA directed against Thra exon 9. Plasmid DNA was injected into fertilized oocytes with a mixed genetic background. Mosaic mice carrying Thra mutations were identified by polymerase chain reaction (PCR)/Sanger sequencing of Thra exon 9 and mated to C57BL/6 mice to derive four mouse lines. In the following, a simplified nomenclature was use for the designation of these four new alleles: ThraS1 = ThraE395fs401X , ThraS2 = ThraE395fs406X , ThraL1 = Thra395fs485X , ThraL2 = ThraK389fs479X . The corresponding proteins are designated as TRα1S1, TRα1S2, TRα1L1, and TRα1L2. The ThraN359Y mutation was generated using the homology-directed repair pathway and designated as ThraM . C57BL/6 fertilized oocytes were microinjected with a solution containing Cas9 mRNA (TriLink BioTechnologies, San Diego, CA), a single-stranded oligonucleotide as a template, and a single-guide RNA prepared by in vitro transcription from a PCR-amplified DNA fragment (Megashort T7 transcription kit; Life Technologies, Courtaboeuf, France). The ThraSlox mutation was generated using a cloning free procedure to modify the TRαAMI locus by the non-homologous end joining pathway. The encoded protein, TRαS3, is almost identical to TRαS1, but the expression context is the same as in TRαAMI/+ mice (see Results). A dual guide RNA was prepared by combining a synthetic trans-activating crRNA 72-mer (tracrRNA) with 3′ modifications for stability (TriLink BioTechnologies), and a synthetic customized crRNA 42-mer with 5′ modifications (Eurogentec, Angers, France). Equimolar quantities of the two RNAs were annealed (2 min at 80°C and 10 min at 37°C, 0.5 μg/μL) and then mixed with an equal volume of Cas9 recombinant protein (1 μg/μL; PNA Bio, Inc., Newbury Park, CA; 10 min at room temperature). The protein/RNA solution was diluted (1/10) in RNAase-free buffer (Tris-HCl 10 mM; EDTA 0.1 mM pH 8; Life Technologies) before microinjection into in vitro fertilized TRαAMI/+ oocytes (16). The final concentrations in the microinjected solution were 50 ng/μL for Cas9 protein and 25 ng/μL for the dual guide RNA. F0 mosaic animals born after reimplantation of microinjected embryos were genotyped by PCR/Sanger sequencing and then mated to C57BL/6 mice to provide F1 founder mice. At least one additional back-cross was performed with C57BL/6 mice before phenotyping in order to avoid possible confounding effects due to off-target mutations. Prediction of sites for off-target mutations, based on sequence similarity, were made with CRISPOR and identified main candidate loci (20). Only one site had fewer than four mismatches with the guide RNA sequence. DNA sequencing was used to search for off-target mutations at 14 of these sites in F0 mice, which were not found (detection limit 10% of genomic DNA; Supplementary Table S1).
Mouse phenotyping
Skeletal analysis was performed by alizarine staining (21). Femur, tibiae, and tail images were recorded under a stereomicroscope (Leica M205 FA). Lengths were measured using Image J software. Micro-computer tomography (CT) X-ray microtomography was performed, as described (22). Histological and immunohistochemical analyses of intestine and cerebellum were as described (23,24). Intestinal progenitor cell proliferation was assessed using anti-Ki67 immunostaining (antibody and fluorescent-conjugated anti-rabbit antibody from Labvision Thermo Fisher Scientific, Courtaboeuf, France) and 4,6-diamidino-2-phenylindole (DAPI) for nuclei counter-staining. Serum levels of interleukin (IL)-1β were measured by enzyme-linked immunosorbent assay (ELISA; Quantikine ELISA Kit; R&D Systems, Minneapolis, MN). Total RNA (Qiagen RNeasy Mini Kit; Qiagen, Hilden, Germany) was extracted from several tissues homogenized in lysis buffer with ceramic beads (Precellys; Bertin Instruments, Montigny-le-Bretonneux, France) for gene expression analysis. RNA (1 μg of each sample) was hybridized to 6-oligomer random primers before reverse transcription (Moloney murine leukemia virus reverse transcriptase; Promega, Charbonnières-les-Bains, France). Two microliters of diluted (1/20) reaction product was used for quantitative PCR (performed in triplicate; SYBR Green iQ supermix with CFX96 real-time PCR machine; Bio-Rad Laboratories, Hercules, CA). Expression levels were calculated using the 2−ΔΔ(Ct) method (25) using Hprt as the reference gene.
In vitro analysis of TRα1 mutations
Wild-type and mutant TRα1 cDNA were amplified from mouse tissues by reverse transcription (RT)-PCR (oligonucleotides in Supplementary Data) and cloned in the pSG5 expression vector (Agilent-Stratagene, San Diego, CA). An expression vector for TRα1N359Y was prepared by using site-directed mutagenesis to modify pSG5-TRα1. Transactivation and dominant-negative properties of mutants TRα1 were assessed in HEK293 cells transfected using TransIT®-LT1 (MirusBio, Madison, WI). Transfected constructs were pSG5 derivatives, pSG5-DR4Luc, in which two DR4 response elements are located upstream to a minimal transcription promoter driving firefly luciferase expression and pBK-βGal as internal control of transfection efficiency. Restriction fragments from pSG5-derived constructs were transferred in pBK-VP16-TRα1 to construct expression vectors encoding VP16-TRα1 fusion proteins with C-terminal mutations. The capacity of VP16-TRα1 proteins to interact with Gal4-NcoR was addressed in a two-hybrid assay in HEK293 cells, using Gal4REx5bglob-luc ( = UAS Luc) as a reporter (26). All luciferase assays were performed in triplicate.
Results
Generation of germline Thra mutations by CRISPR/Cas9 genome editing
Two different strategies were used to generate targeted Thra mutations in the mouse germline by CRISPR/Cas9 genome editing. The first strategy consisted of microinjecting mouse oocytes with a DNA construct to produce transiently both Cas9 nuclease and a single guide RNA directed to exon 9 (19). This introduced a double-strand break in genomic DNA, which was repaired by non-homologous end joining, an error-prone mechanism. Embryo reimplantation produced several mosaic founders (F0). These were identified by PCR/Sanger DNA sequencing. Unambiguous mutation identification was achieved after germline transmission to F1 animals, which eliminates mosaicism. Four frameshift mutations (E395fs401X, E395fs485X, E395fs406X, and K389fs479X) were selected for the generation of new mouse lines. E395fs401X and E395fs406X mutations are +1 frameshifts, introducing stop codons few nucleotides downstream to the mutated codon. This corresponds to the frameshift mutations found in some RTHα patients. In particular, the E395fs406X mutation is very similar to the F397fs406X human mutation (27). The two other mutations were +2 frameshifts, which have no known equivalent in human patients, resulting in the production of an amphigoric protein (28) with a 90 amino acid C-terminal extension. These four frameshift mutations are not predicted to alter corepressor interaction, mapped to the intact part of the ligand binding domain (29), but eliminate the C-terminal helix 12 required for coactivator recruitment (30) (Fig. 1). A second strategy was used to produce a fifth mutant line, using the homology-directed repair pathway. In that case, site-directed mutagenesis was achieved by microinjecting mouse oocytes with the Cas9 mRNA and a single guide RNA together with a single-stranded DNA template (99-mer) serving as template. PCR/Sanger DNA sequencing was then used to identify the designed mutation (N359Y), which is identical to the one reported for a human patient with atypical presentation (5). There is no frameshift in this model. Therefore, apart from the N359Y amino acid substitution, the C-terminal part of TRα1, including helix 12, is intact (Fig. 1). The N359Y amino acid substitution reduces the affinity of TRα1 for T3, but does not alter the interface required for coactivator or corepressor recruitment (5). In the following, the simplified nomenclature will be used described in the Methods section: ThraS1 , ThraS2 , ThraL1 , ThraL2 , and ThraM (S for shorter, L for longer, and M for missense). The corresponding proteins will be designated as TRα1S1, TRα1S2, TRα1L1, TRα1L2, and TRα1M.

Using CRISPR/Cas9 gene edition to generate mutations in exon 9 of the mouse Thra gene. (
Overall phenotype
Phenotypes were analyzed on F2 heterozygous animals and later generations in order to ensure genetic segregation between the identified Thra mutations and other possible off-target mutations created by the Cas9 nuclease. All controls were littermates, with an identical genetic background. As significant literature is already available on the consequences of Thra knock-out and knock-in mutations in mice (31,32), several traits were selected in bone, brain, blood, heart, and intestine for phenotypic characterization. The choice was based on the ease of quantitative analysis, putative discrepancies with previous mouse models, and relevance to the main clinical observations made in patients with RTHα. The overall phenotype of mutant mice was close to normal (examples for TRα1S1/+ mice are shown on Fig. 2). Mutations had no visible effect on viability and fertility. Although there was a general trend toward lower body weight, the weights of most adults remained close to normal range (Table 1).
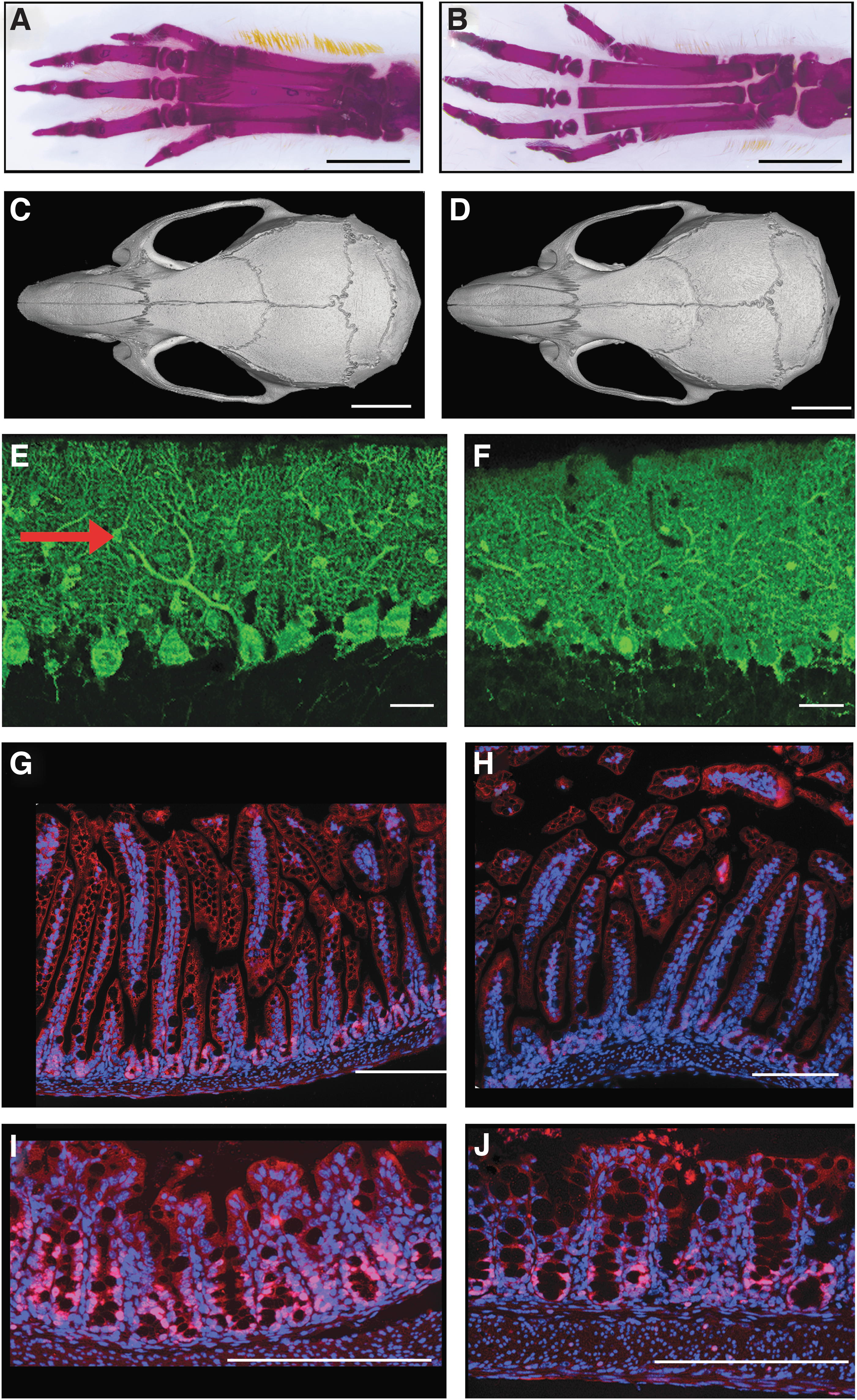
Phenotype of ThraS1/+
mice. The control is on the left side, the mutant on the right. (
Statistically significant values are shown in bold.
p < 0.05; ** p < 0.01.
Skeletal phenotype
Whole-mount alizarine staining of skeletons, performed on postnatal day 15 (PND15), indicated retarded ossification in long bones (Fig. 2A and B), with persistence of thick cartilage plates at the extremities, for all mutant lines except ThraM/+ . This defect was easily visible in the tail, in particular for ThraS1/+ and ThraS2/+ mice (Fig. 3). Adult femur lengths were significantly decreased in ThraS1/+ and ThraS2/+ male and female mice but not in the other lines (Table 1). Interestingly, this reduction in size did not affect all long bones, as no reduction was noticed for tibiae (data not shown). X-ray microtomography analysis of adult femurs also revealed defects in bone microstructure. A reduction in trabecular thickness and an increase in trabecular bone surface was found in ThraS1/+ , ThraS2/+ , and ThraM/+ but not in ThraL1/+ or ThraL2/+ mice (Table 1). These may be signs of osteoporosis, although all other bone microstructure parameters remained within normal range. Careful examination of skeletons, after whole-mount alizarine staining of adults and juveniles, failed to reveal any bone malformation. X-ray microtomography was performed on the skull of a small set of adult ThraS1/+ males (Fig. 2C and D). This analysis did not reveal any bone thickening or presence of Wormian bones, two phenotypes that were reported in several RTHα patients. In summary, ThraS1/+ and ThraS2/+ mice displayed alterations in bone maturation, growth, and microstructure that mirror the skeletal defects reported in patients with RTHα. These skeletal defects were less pronounced or absent in ThraL1/+ and ThraL2/+ mice. Only bone microstructure was altered in ThraM/+ mice (Table 1).
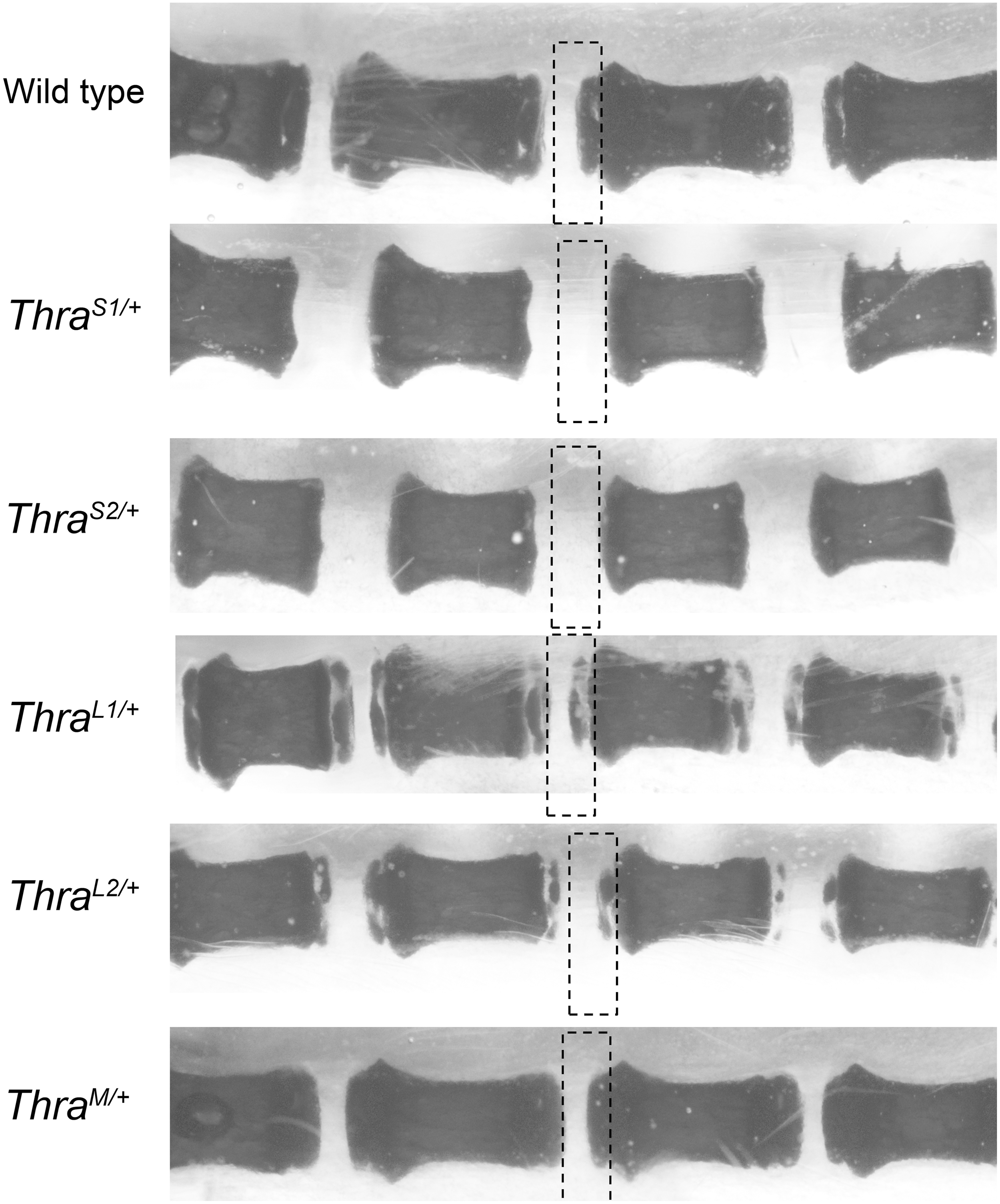
Thra mutations delay postnatal ossification of tail vertebrae. Alizarine staining of tail vertebrae (PND 15). The rectangles indicate the position on one vertebra extremity where secondary ossification is expected to take place. Secondary ossification is complete in wild-type mice. Most cartilage, unstained, has been replaced by ossified bone. Secondary ossification did not start in ThraS1/+ and ThraS2/+ mice and is partial in ThraL1/+ and ThraL2/+ mice. ThraM/+ mice do not visually differ from wild-type mice.
Blood and heart phenotype
A modest increase in the serum level of free T3 was found in ThraS1/+ and ThraS2/+ mice only, resulting in a significant decrease in the free T4/free T3 ratio, as in most RTHα patients. Blood analysis revealed a slight decrease in hemoglobin content only in ThraS1/+ and ThraS2/+ mice (Table 1). Spleen size on PND15 was not changed, arguing against a major defect in postnatal erythropoiesis (33). The serum level of IL-1β was measured in ThraS1/+ adult mice and control littermates. IL-1β was not detectable (detection limit 10 pg/mL; seven wild-type males, seven wild-type females, six ThraS1/+ males, and six ThraS1/+ females), arguing against the occurrence of chronic inflammation. Serum calcium levels were measured in the blood of ThraM/+ mice, searching for the dysregulation of calcium homeostasis reported in the corresponding human patient (5), but a major change was not observed (10.6 ± 0.1 mg/dL, n = 8 vs. 10.9 ± 0.1 mg/dL, n = 10, in wild-type littermates; p = 0.06). In order to evaluate the influence of the mutations in the heart, RT-qPCR was used to measure the cardiac expression of Hcn2, a known TRα1 target gene in cardiomyocytes (Table 1). A marked reduction in Hcn2 expression was found in ThraS1/+ and ThraS2/+ mice. The decrease was modest in ThraL2/+ and not significant in ThraM/+ and ThraL1/+ mice.
Neurodevelopment
Cerebellum histology failed to reveal typical signs of hypothyroidism in mutant mice. In particular, parvalbumin immunostaining on PND21 did not reveal any obvious alteration in the arborization of Purkinje cells or their alignment as a single cell layer (Fig. 2E and F). The density of parvalbumin-immunoreactive cells in the molecular layer was also measured to address a possible delay in the differentiation of basket and stellate cells (34). A modest diminution was visible when all mutant mice were considered together (relative density: wild type: 1.00 ± 0.29, n = 15; mutants: 0.79 ± 0.28, n = 16; p < 0.05). However, this was only a minor effect, which did not reach a significance threshold when individual mutant groups were considered. This defect was not associated with any visible accumulation of immature Pax2+ basket and stellate cell progenitors (24). As Hr and Klf9 are two genes known to be T3-responsive in the cerebellum, RT-qPCR was used to measure the levels of the corresponding mRNA. A major decrease in Hr expression was observed mainly in ThraS1/+ and ThraS2/+ mice, while a modest but significant reduction was found in ThraL2/+ mice (Table 1). There was also a significant reduction in Klf9 mRNA levels in the cerebellum of ThraS1/+ and ThraS2/+ but not in the other mice. Taken together, these analyses indicate a moderate hypothyroid-like phenotype in the cerebellum, most obvious in ThraS1/+ and ThraS2/+ mice.
Intestinal epithelium
Histological examination of the small intestine and colon was performed without noticing any evident ultrastructural alterations (Fig. 2G–J). Ki67 immunostaining was also performed to label actively proliferating epithelial cells, and a noticeable reduction in cell proliferation, in the colon and ileum, was found, as previously reported for several Thra knock-out models (35,36). Due to small sample size, the diminution did not always reach a significance threshold (Table 1).
Phenotype severity correlates with strong and permanent interaction with corepressor
A striking general conclusion of the above analyses is that the phenotypes of ThraS1/+ and ThraS2/+ mice are more pronounced than the ones of ThraL1/+ , ThraL2/+ , and ThraM/+ , for which many developmental and physiological parameters remain within normal range. Further in vitro experiments were performed after cloning all Thra cDNA to evaluate the molecular basis of the peculiar severity of the ThraS1/+ and ThraS2/+ phenotypes. A transient expression assay confirmed that among the five mutations under study, only TRα1M, expressed in ThraM/+ mice, possesses some residual transactivation activity (Fig. 4A). These results confirm that coactivator recruitment by the C-terminal helix 12 (present in TRα1M but not in the other proteins) is mandatory for transactivation and provides a likely explanation for the attenuated phenotype of ThraM/+ mice. When cotransfected with intact TRα1, only TRα1S1 and TRα1S2 exerted a clear dominant-negative activity (Fig. 4B). Western blotting, performed on transfected cells, showed equivalent expression levels for the mutant proteins, arguing against the possibility that C-terminal changes modified the protein stability (Supplementary Fig. S1). Immunoprecipitation of tagged receptors was also performed after DNA transfection to evaluate the capacity of the mutations to alter interactions between TRα1 and cellular proteins in an unbiased manner. This survey revealed that the interaction of a number of proteins with the intact receptor depends on the presence of T3. It also illustrated the inability of TRα1S1, TRα1S2, TRα1L1, and TRα1L2 receptors to recruit factors in a T3-dependent manner, presumably due to the absence of helix 12. This analysis did not provide any clue for the understanding of the different outcomes associated with the four frameshift mutations (Supplementary Fig. S2). Thus, a two-hybrid assay (4) was used to evaluate precisely the capacity of each mutant receptor to interact with NcoR, as this transcription corepressor has been shown to mediate a significant part of the detrimental effects of TRα1 mutations in mice (37). NcoR interaction with wild-type TRα1 and TRα1M was progressively destabilized by increased concentrations of T3, confirming that the N359Y substitution does not abolish the receptor response to T3 (Fig. 4C). By contrast, the absence of the terminal-helix in TRα1S1, TRα1S2, TRα1L1, and TRα1L2 eliminates the capacity of T3 to destabilize the NcoR interaction with the receptor. Importantly, the shortened proteins (TRα1S1 and TRα1S2) presented a stronger interaction with NcoR than the extended proteins (TRα1L1 and TRα1L2).
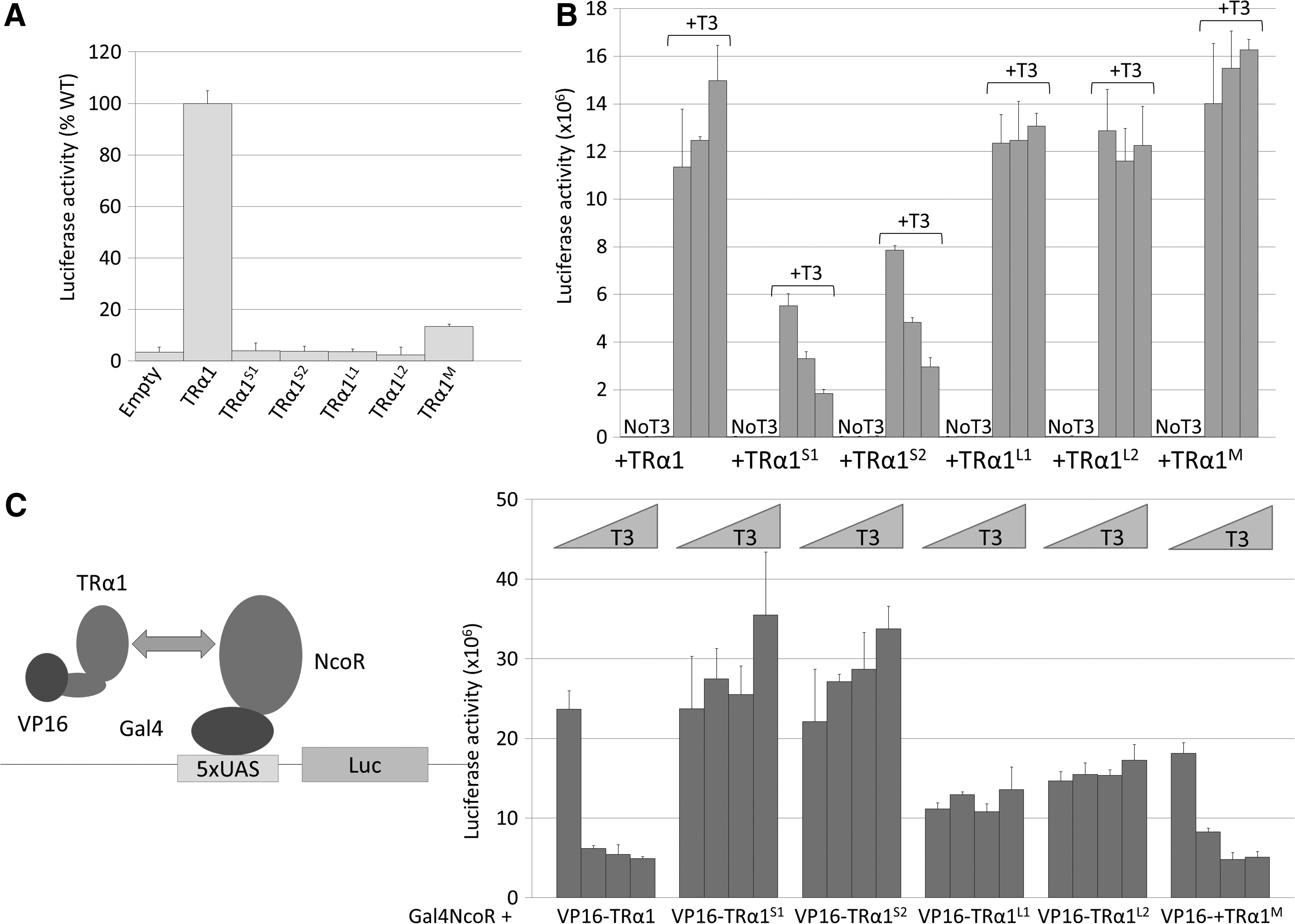
Consequences of TRα1 mutations on receptor function. (
Increasing the expression level of altered receptors exacerbates disease severity
The relatively mild phenotypes of the five mutant mouse models presented here contrasts with the phenotypes previously reported for mice heterozygous for Thra knock-in mutations, in which several unique traits were found. Notably, lethality within 3 weeks after birth was previously reported for TRαAMI/+ mice, whether TRα1L400R is expressed in all tissues (16) or only in the brain (38). This mutation, located in helix 12, provokes major neurodevelopmental defects (24,38,39). However, the TRαAMI allele results from an extensive remodeling of the Thra gene, which eliminates all the other Thra encoded proteins, to encode TRα1L400R exclusively (Fig. 5A), making comparison with CRISPR/Cas9-generated models difficult. In order to compare the consequences of a frameshift mutation and the L400R substitution, CRISPR/Cas9 editing was performed on the TRαAMI/+ mouse embryo. The single guide RNA, used previously to generate frameshifts, was introduced in TRαAMI/+ fertilized oocytes to generate mutations upstream to codon 400 (Fig. 5B). This editing introduced an additional frameshift mutation in TRαAMI , which eliminated the C-terminus of the receptor (Fig. 5B). In the resulting allele, called ThraSlox/+ , the encoded protein, TRαS3, is almost identical to TRαS1, but the expression context is the same as in TRαAMI/+ mice. ThraSlox/+ were mated to NestinCre mice to express the modified receptor in the brain. RT-PCR and Sanger sequencing were then combined to compare the relative levels of mutant and wild-type Thra mRNAs precisely in heterozygous mice. Peak surfaces provided an indication of the respective mRNA abundance in postnatal cerebellum. Allelic balance was in favor of wild-type mRNAs in ThraS1/+ mice, while the opposite was found in ThraSlox/+xNestinCre and TRαAMI/+xNestinCre mice (Fig. 5C). Both genotypes were associated with postnatal mortality (n = 6) and obvious histological defects in the cerebellum (data not shown). Overexpression of the mutant allele most likely results from the elimination of alternate splicing from the Thra locus, the transcription of which is entirely dedicated to TRα1L400R or TRα1S3 mRNA synthesis. This overexpression is likely to be the predominant cause of an aggravated phenotype.
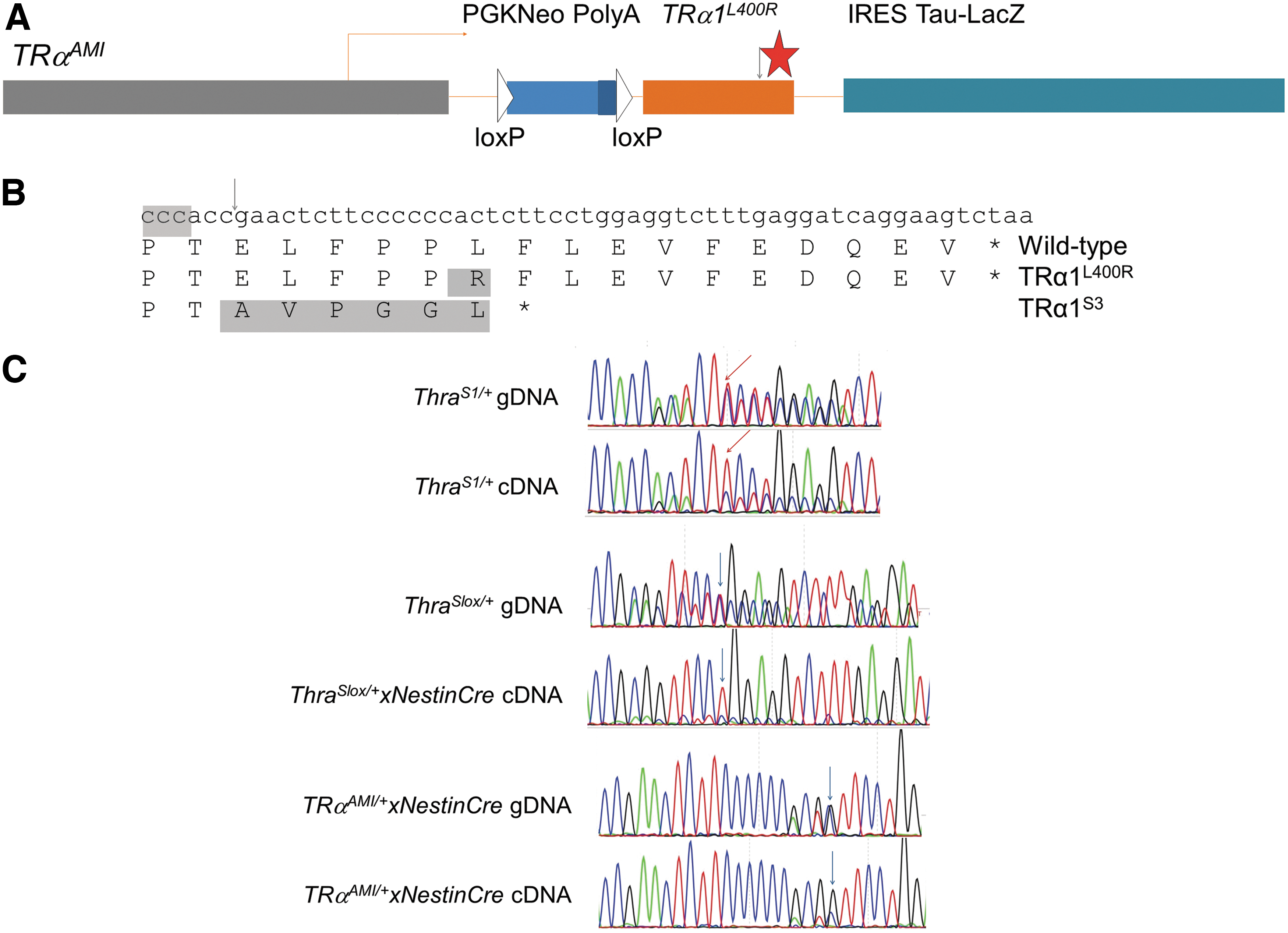
Elimination of alternate splicing leads to overexpression of TRα1S1 in brain cells. (
Discussion
Advantage was taken of the recently developed CRISPR/Cas9 methodology to generate five new mouse models of the RTHα syndrome. This includes a model customized to match the missense mutation encoding TRα1N359Y discovered in a patient with uncharacteristic symptoms. Compared to the use of homologous recombination in embryonic stem cells, which previously provided four mouse models (14 –17), and in addition to its efficiency and simplicity, genome editing offers two key advantages. First, it is performed without introducing superfluous genetic modifications that can alter the expression level of Thra-encoded proteins. Second, it helps to produce animal models with identical genetic backgrounds, permitting precise side-by-side comparisons of different mutations. A current limitation of the method is that it also generates off-target mutations. The absence of off-target mutations were verified at several genomic positions, predicted as sensitive by bioinformatics mean. It is, however, known that such predictions are not fully reliable (40), and the presence of off-target mutations in founder mice cannot be ruled out. Provided that they are located at sufficient chromosomal distance to the targeted site, these off-target mutations quickly segregate during germline transmission. Since phenotyping was started after at least two germline transmissions, and as littermates were used as controls, these off-target mutations are not expected to bias the conclusions. They might, however, generate some phenotypic variability within each group of mice. The CRISPR/Cas9-generated mutations of Thra caused phenotypic alterations resembling the ones reported in human patients: impaired skeletal growth, mild alterations of neurodevelopment, tendency toward anemia, and reduction of the T4/T3 ratio. No signs of chronic inflammation were found, unlike what has been reported after Thra knock-out (41). Also, some traits repeatedly reported in human RTHα patients were not observed in mice. These include severe constipation, increased thickness of the skull, and bone malformations.
This study is probably one of the first to illustrate the promising approach of modeling human sporadic cases of a genetic disease in mice in order to address the causal relationship between a mutation and a unique phenotype. This approach greatly clarifies the phenotypic consequence of the TRα1N359Y missense mutation present in a single patient (5). Except for the short stature, none of this patient's atypical traits, notably hypercalcemia, macrocytic anemia, diarrhea, absence of clavicles, and bone deformations, could be reproduced in mice. These alterations are believed to result from either a nongenetic cause or from mutations located in non-coding regions.
The current observations also shed some light on divergent observations made previously on the first four Thra knock-in mouse models, which were produced several years before the discovery of RTHα (42). In particular, evidence is provided that the postnatal lethality of TRαAMI/+ mice, expressing the TRα1L400R mutation, results from an increased expression of the mutant allele compared to the wild-type allele. This does not rule out that the L400R amino acid substitution might have unique consequences on the receptor function, which would enhance its dominant-negative capacity. In particular, previous in vitro investigations showed that TRβ1L454R, equivalent to TRα1L400R in the TRβ1 context, has the peculiar capacity to eliminate coactivator recruitment completely (30). Considering the allele design, it can be predicted that overexpression of mutant TRα1 also occurs in two other Thra knock-in models, TRα1R384C/+ and TRα1PV/+ , in which alternate splicing is eliminated as well. It is hypothesized that this artificial overexpression leads to exaggerated phenotypic manifestations. This conclusion reinforces the view according to which alternate splicing of Thra transcripts may serve to limit TRα1 expression, besides producing TRα2, a protein whose putative function remains elusive (43). Overall, CRISPR/Cas9-generated murine mutations prove to be more relevant models of the human RTHα than available knock-in models.
This side-by-side phenotype analysis also firmly establishes that seemingly equivalent TRα1 mutations lead to phenotypes with different degrees of severity. Interestingly, it was found that disease severity correlates with the ability of the mutant receptor to interact with NcoR corepressor, even in the presence of T3. In particular, it seems that the protruding C-terminus of TRα1L1 and TRα1L2 hinders the interaction with NcoR. This suggests that any pharmacological intervention that would destabilize this interaction could be beneficial to RTHα patients.
Footnotes
Acknowledgments
We thank M. Privalsky, K. Chatterjee, and Maura Agostini for the plasmid gift, and C. Le Nevé, A. Claret, and E. Ravel for help with the analysis. We thank the facilities of SFR Biosciences (Lyon, France) for excellent technical assistance: X-ray microtomography (AniRA-ImmOs), microscopy (PLATIM), mouse breeding and transgenesis (AniRA-PBES), and Anipath facility for histology (SFR Santé Lyon Est). This work was supported by grants from Agence Nationale de Recherche (Thyromut2 program; ANR-15-CE14-0011-01) and from Fondation ARC pour la Recherche sur le Cancer to M.P. (Programme Labellisé PGA1201402000834).
Author Disclosure Statement
The authors have nothing to declare.