
Abstract
Background:
Primary congenital hypothyroidism (CH) affects about 1:3000 newborns worldwide and is mainly caused by defects in thyroid gland development (thyroid dysgenesis [TD]) or hormone synthesis. A genetic cause is identified in <10% of TD patients. The aim was to identify novel candidate genes in patients with TD using next-generation sequencing tools.
Patient Findings:
Whole exome sequencing was used to study two families: a consanguineous Tunisian family (one child with severe thyroid hypoplasia) and a French family (two newborn siblings, with a thyroid in situ that was not enlarged on ultrasound at diagnosis). Variants in candidate genes were filtered according to type of variation, frequency in public and in-house databases, in silico prediction tools, and inheritance mode. Unexpectedly, three different variants of the thyroid peroxidase (TPO) gene were identified. A homozygous missense mutation (c.875C>T, p.S292F) was found in the Tunisian patient with severe thyroid hypoplasia. The two French siblings were compound heterozygotes (c.387delC/c.2578G>A, p.N129Kfs*80/p.G860R) for TPO mutations. All three mutations have been previously described in patients with goitrous CH. In these patients, treatment was initiated immediately after diagnosis, and the effect, if any, of thyrotropin stimulation of these thyroids remains unclear.
Conclusions:
The first cases are reported of thyroid hypoplasia at diagnosis during the neonatal period in patients with CH and TPO mutations. These cases highlight the importance of screening for TPO mutations not only in goitrous CH, but also in normal or small-size thyroids, and they broaden the clinical spectrum of described phenotypes.
Introduction
C
Patients
Family 1
A male (patient #1) born to consanguineous parents (first cousins) from Sfax, Tunisia, was diagnosed with CH at day 10 of life, with a thyrotropin (TSH) level of 52 mIU/L (normal range [N] 0.7–10.1 mIU/L) and a free thyroxine (fT4) level of 14.2 pmol/L (N 9.3–23.5 pmol/L). Thyroid ultrasound (US) showed a non-goitrous eutopic thyroid, confirmed by scintigraphy. Levothyroxine (LT4) treatment was initiated immediately. The patient had a normal growth and puberty pattern, normal cognitive development, and mild bilateral sensorineural hearing loss. At 25 years of age, the patient was treated with LT4 at a dose of 200 μg/day (2.2 μg/kg/day), and he had a TSH of 3.16 mIU/L. The thyroid US showed hypoplasia, with a volume of 1.3 mL (N 12 ± 5 mL). At 27 years of age, LT4 was withdrawn for 49 days, and a thyroid US performed after withdrawal (TSH >100 mIU/L (N 0.27–5 mIU/L) and fT4 < 0.3 pmol/L (N 12–22 pmol/L) confirmed thyroid hypoplasia (2.9 mL; N 12 ± 5 mL). Scintigraphy showed normal 99mTc uptake by the small normally located thyroid. Thyroid US and thyroid function tests were normal in the two parents and two other siblings (Fig. 1A).
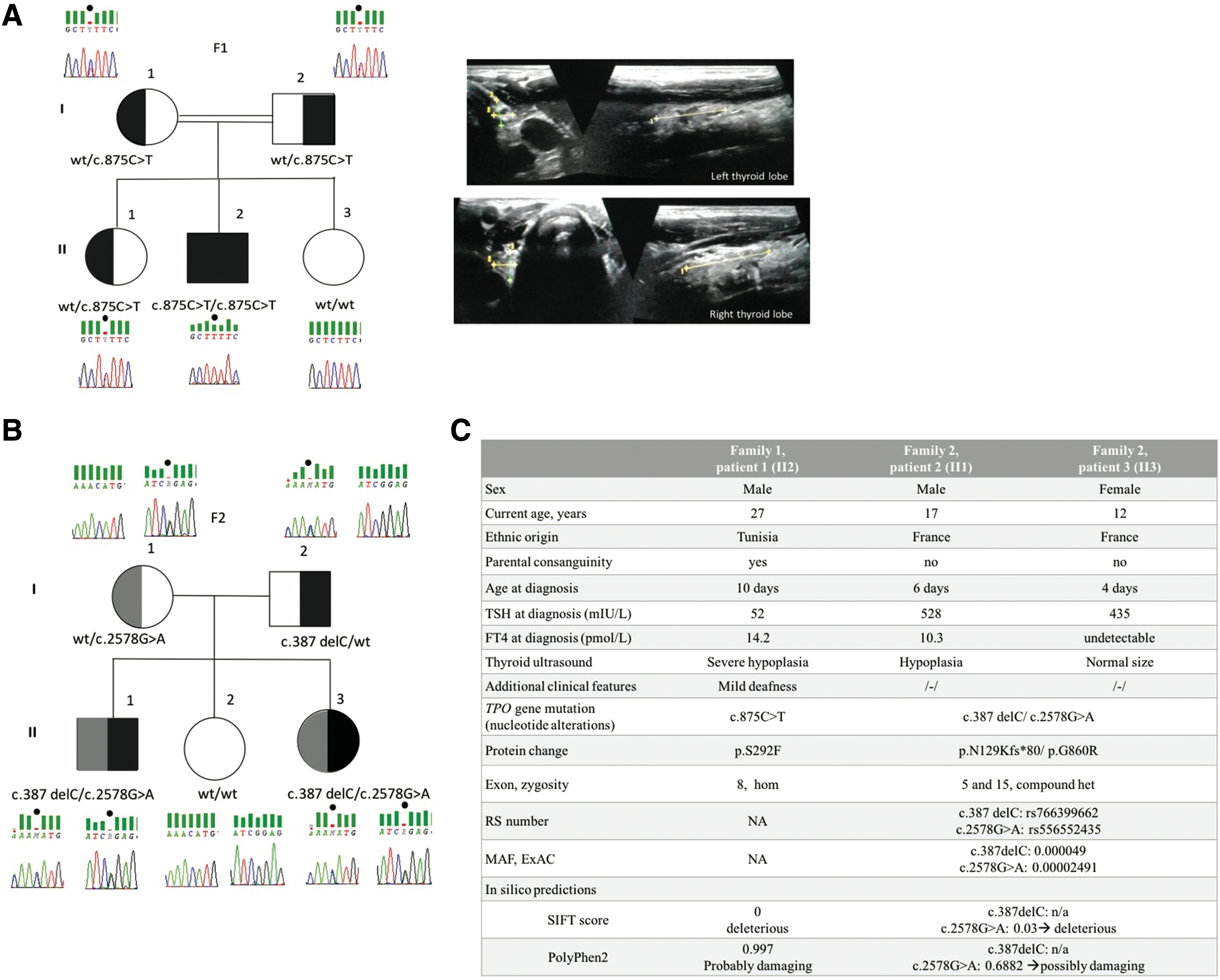
Pedigrees of the two families harboring TPO mutations and clinical characteristics of described patients. (
Family 2
The 17-year-old boy (patient #2) and his 12-year-old sister (patient #3) born to nonconsanguineous French parents were both diagnosed with CH by routine neonatal screening. CH was confirmed within a few days by serum tests showing TSH elevation (528 mIU/L and 435 mIU/L in patients #2 and #3, respectively) and low fT4 levels (10.3 pmol/L and undetectable, respectively). At diagnosis, thyroid US showed a thyroid in situ, without enlargement. The parents and the third sibling had normal thyroid function tests. Patient #2 had a small thyroid on US performed during follow-up, under LT4 and normal TSH values. At reevaluation, thyroid US showed thyroid hypoplasia in patient #2 at 14 years of age and a normal-sized gland in situ in patient #3 at nine years of age, with thyroid volumes of 5 and 6.6 mL, respectively (N 8 ± 3 mL). These US findings were confirmed by another thyroid US at 17 and 12 years of age for patients #2 (thyroid volume of 4.5 mL) and #3 (thyroid volume of 6.5 mL), respectively (Fig. 1B).
WES and data analysis
All patients, parents, and siblings gave written informed consent for participation in the study and genetic testing. Genomic DNA was isolated from whole blood using the QIAmp DNA Blood Mini kit (Qiagen, Courtaboeuf, France) according to the manufacturer's instructions. WES data analysis and filtering of variants were performed, as previously described (detailed in the Supplementary Data; Supplementary Data are available online at
Results
Three variants (with allele frequencies <1%) in the TPO gene (RefSeq transcript NM_000547.5) were found by WES. Patient #1 was homozygous for the missense mutation c.875C>T, p.S292F in exon 8 (5). Both parents and one sibling were carriers. The in silico assessment (SIFT,
Discussion
Biallelic TPO mutations are known to cause CH with thyroid in situ and most frequently goiter, which may develop during the fetal/neonatal period or at any time in childhood (8). Here, patients harboring TPO mutations with unexpected thyroid hypoplasia at birth are reported.
TPO encodes for thyroid peroxidase, located on the apical thyrocyte membrane and participating in thyroid hormonogenesis. TPO mutations impair thyroid function by impeding iodine organification, thereby leading to thyroid gland enlargement through chronic TSH stimulation (9). All three variants reported here have been published in patients with dyshormonogenesis (5 –7) and are in highly conserved and functional TPO domains. The biallelic mutation in exon 8, p.S292F, has been associated with sensorineural hearing loss (as a consequence of the CH), a feature in patient #1, and with multinodular goiter and follicular carcinoma (5). The p.N129Kfs*80 variant may be responsible for the loss of a crucial region for TPO function (6). Finally, exon 15 encodes the membrane-spanning part of the enzyme, and the nucleotide variation in patients #2 and #3 (c.2578G>A, p.G860R) is likely to impair insertion of the TPO protein into the apical membrane (7). The combination of these two TPO variants is likely to result in altered TPO function and CH.
To the authors' knowledge, the patients reported here are the first to have TPO mutations and small thyroid glands. These unexpected findings are consistent with recent data on SLC26A4/PDS (10) and DUOX2 (11) mutations in patients with CH phenotypes presenting with hypoplasia and non-goitrous hypothyroidism. Furthermore, in a previously described rat model, WIC-rdw rats have dwarfism and non-goitrous hypothyroidism due to a missense mutation in the Tg gene (12). The thyroid is hypoplastic in rdw/rdw rats, in contrast to previously reported Tg mutations. A suggested hypothesis to explain the absence of goitrogenesis may be the cytotoxic effect of the misfolded protein (13). This could be a hypothesis for hypoplasia in the patients in this study. However, it does not explain why previously described patients harboring the same mutation presented with a goiter. A second additional genetic or epigenetic hit in the thyroid tissue could explain the small and not enlarged thyroid in these patients at birth (14). Moreover, in these patients, treatment was initiated immediately after diagnosis, and it is unclear whether their thyroid glands would have responded and have grown under postnatal TSH stimulation. TPO is expressed at the late stages of thyroid development, making a primary structural defect unlikely. It is also noteworthy that WES did not identify any variants in genes involved in defects associated with TD such as TSHR, NKX2-1, PAX8, FOXE1, GLIS3, BOREALIN, or NKX2-5. A genetic cause is known only for about 10% of TD cases. In a large cohort of CH patients screened by a next-generation sequencing approach, including 11 TD and dyshormonogenesis genes, one or more disruptive variants were found for only 32.9% of patients, confirming that the genetic background of TD with a gland in situ still remains poorly understood and may have a complex etiology (15). Conceivably, additional epigenetic or genetic factors (modifier genes) and environmental influences (endocrine disruptors, iodine status) may also influence the functional and structural phenotype.
In conclusion, patients with CH harboring TPO mutations with unexpected thyroid hypoplasia at birth are described. However, it remains elusive with how these biallelic TPO mutations explain the hypoplastic phenotype and additional (epi)genetic or non-genetic mechanisms, not yet unraveled, may also be required.
Footnotes
Acknowledgments
We thank the patients and families for their kind participation. This study was partly funded by a grant from the nonprofit organization Fondation Maladies Rares. A.S. was supported in part by grants from the European Society for Pediatric Endocrinology Research Fellowship, Belgian Kids Fund for Pediatric Research, French Pediatric Endocrinology Society, and Onassis Foundation. A.C., M.G., and M.P. were supported by EDF, SANDOZ SAS, Merck Serono France, and the Princess Grace Foundation of Monaco. We are also deeply grateful to the Imagine Institute Biobank, Paris, France.
Author Disclosure Statement
The authors report no conflicts of interest in this work.