
Abstract
Background:
3-Iodothyroacetic acid (TA1) is among the thyroid hormone (T3) metabolites that can acutely modify behavior in mice. This study aimed to investigate whether TA1 is also able to reduce neuron hyper-excitability and protect from excitotoxic damage.
Methods:
CD1 male mice were treated intraperitoneally with saline solution or TA1 (4, 7, 11, or 33 μg/kg) before receiving 90 mg/kg pentylenetrazole subcutaneously. The following parameters were measured: latency to first seizure onset, number of mice experiencing seizures, hippocampal levels of c-fos, and PI3K/AKT activation levels. Organotypic hippocampal slices were exposed to vehicle or to 5 μM kainic acid (KA) in the absence or presence of 0.01–10 μM TA1. In another set of experiments, slices were exposed to vehicle or 5 μM KA in the absence or presence of 10 μM T3, 3,5,3′-triiodothyroacetic acid (TRIAC), T1AM, thyronamine (T0AM), or thyroacetic acid (TA0). Neuronal cell death was measured fluorimetically. The ability of TA1 and T3, TRIAC, T1AM, T0A, and TA0 to activate the PI3K/AKT cascade was evaluated by Western blot. The effect of TA1 on KA-induced currents in CA3 neurons was evaluated by patch clamp recordings on acute hippocampal slices.
Results:
TA1 (7 and 11 μg/kg) significantly reduced the number of mice showing convulsions and increased their latency of onset, restored pentylenetrazole-induced reduction of hippocampal c-fos levels, activated the PI3K/AKT, and reduced GSK-3β activity. In rat organotypic hippocampal slices, TA1 reduced KA-induced cell death by activating the PI3K/AKT cascade and increasing GSK-3β phosphorylation levels. Protection against KA toxicity was also exerted by T3 and other T3 metabolites studied. TA1 did not interact at KA receptors. Both the anticonvulsant and neuroprotective effects of TA1 were abolished by pretreating mice or organotypic hippocampal slices with pyrilamine, an histamine type 1 receptor antagonist (10 mg/kg or 1 μM, respectively).
Conclusions:
TA1 exerts anticonvulsant activity and is neuroprotective in vivo and in vitro. These findings extend the current knowledge on the pharmacological profile of TA1 and indicate possible novel clinical use for this T3 metabolite.
Introduction
T
Aberrant neuronal discharge, secondary to increased brain levels of excitatory mediators, is among the causes of convulsions and a trigger for excitotoxic-induced cell death. From a therapeutic point of view, different classes of anticonvulsant drugs are available, but it remains to be determined whether these drugs are effective in protecting neurons against excitotoxicity (10).
Considering that TA1 could potentially represent an active metabolite of T3 and of T1AM, this study sought to investigate whether TA1 also has neuroprotective activity in respect of drug-induced convulsions and excitotoxic neuron death. This aim was explored in mice treated with a single subcutaneous (s.c.) dose of pentylenetetrazole (PTZ), a selective GABA-A receptor antagonist, a treatment that leads to neuronal hyper-excitability (11), and in organotypic hippocampal slices exposed to kainic acid (KA), a well known model to study drug-induced neuroprotection (12 –14).
The PTZ model investigated whether TA1 treatment (i) protects mice from drug-induced convulsions and (ii) activates signaling in mouse hippocampus, including the PI3K/AKT cascade and conserves c-fos levels. In organotypic hippocampal slices, the study investigated whether TA1 reduced KA-induced neuronal death, if it activates survival mechanisms including the PI3K/AKT cascade, and if TA1 effects are shared by other iodinated and not iodinated T3 metabolites, including T1AM, on excitotoxicity.
To investigate the involvement of the histamine system, the effect of TA1 was also studied in mice pretreated with pyrilamine (PYR), a histamine type 1 receptor (H1R) antagonist, before receiving PTZ, and in organotypic hippocampal slices exposed to PYR before TA1 and before KA challenge.
Methods
Animals
Male CD1 mice (weight 20–30 g) purchased from Envigo were used in the present study. Five mice were housed per cage. Cages were placed in the experimental room 24 h prior to testing to ensure adaptation. Animals were housed at 23 ± 1°C under a 12 h light–dark cycle (lights on at 7:00am) and were fed a standard laboratory diet with ad libitum access to water. Experiments and animal use procedures were in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publications No. 80-23, revised 1996). The experimental protocols were approved by the Ethical Committee of the Italian Council of Health, in compliance with the European Convention for the Protection of Vertebrate Animals used for Experimental and Other Scientific Purposes (ETS no. 123) and the European Communities Council Directive of 24 November 1986 (86/609/EEC). The authors further attest that all efforts were made to minimize the number of animals used and their suffering.
Organotypic hippocampal slice cultures were prepared from male and female Wistar rat pups (7–9 days old) obtained from Charles River. Acute hippocampal slices were prepared from Wistar rats (15–28 days old), which were also obtained from Charles River.
In vivo model
Male CD1 mice received an intraperitoneal (i.p.) injection of vehicle (saline solution) or TA1 (4, 7, 11, and 33 μg/kg) and, after 10 min, 90 mg/kg PTZ s.c. Mice were then individually housed in transparent Plexiglas cages (25 cm × 15 cm × 10 cm) and observed for 30 min. The number of convulsing mice and the latency to first clonic seizure occurrence were recorded. For the purposes of this experiment, clonic seizure activity in mice was defined as clonus of the whole body lasting >3 s, accompanied by loss of the righting reflex (13). At the end of the observation period, animals were then housed in cages, fed a standard laboratory diet with ad libitum access to water, and sacrificed 24 h later. The hippocampi of the mice were removed and stored at −80° until Western blot analysis.
Additional mice were then treated i.p. with 7 μg/kg TA1 or saline (Veh) and sacrificed 24 h later to remove the hippocampi for Western blot analysis.
In another set of experiments, PTZ (90 mg/kg) was administered s.c. to mice that had received either vehicle or the H1R antagonist PYR (10 mg/kg i.p.; Sigma–Aldrich SRL) 20 min before the i.p. injection of vehicle or 7 μg/kg TA1. The number of mice showing convulsions and the latency to first clonic seizure occurrence were recorded, as previously described.
A TA1 stock solution (10 mM) was prepared in 100% dimethyl sulfoxide, and then appropriate dilutions were made in saline solution (14).
Preparation of rat organotypic hippocampal slice cultures
Organotypic hippocampal cultures were prepared, as previously described (12 –14). Briefly, hippocampi were removed from the brains of Wistar rat pups. Transverse slices (420 μm) were prepared using a McIlwain tissue chopper and transferred onto 30 mm diameter semi-porous membrane inserts (Millicell-CM PICM03050; Millipore), which were placed in six-well tissue culture plates containing 1.2 mL of medium per well. The slice culture medium consisted of 50% Eagle's minimal essential medium, 25% heat-inactivated horse serum, 25% Hanks' balanced salt solution, 5 mg/mL glucose, 2 mM L-glutamine, and 3.75 mg/mL amphotericin B. Slices were incubated at 37°C in an atmosphere of humidified air and 5% CO2 for two weeks. Prior to experimentation, all slices were screened for viability by incubating them for 30 min with the fluorescent dye propidium iodide (PI; 5 μg/mL; Sigma–Aldrich SRL). Slices displaying signs of neurodegeneration were excluded from the study.
Slices were exposed to vehicle or 10 μM TA1 for 24 h. At the end of the incubation period, slices were collected for Western blot analysis to evaluate levels of p-AKT, p-GSK-3β, p-mTOR, and phospho-p70S6K. In another set of experiments, slices were exposed for 24 h to 10 μM T3, T1AM, 3,3′,5-triiodothyroacetic acid (TRIAC), thyronamine (T0AM), and thyroacetic acid (TA0). At the end of the incubation period, slices were collected for Western blot analysis to evaluate levels of p-AKT.
Cell toxicity evaluation
Organotypic hippocampal slices are considered appropriate for in vitro analysis of the neuroprotective effects of drugs. A specific neuronal toxicity in the CA3 region is achieved by exposing slices to 5 μM KA for 24 h (Tocris) (12). This model was utilized for different experimental conditions, as described in the following section.
Slices were exposed to vehicle or increasing concentrations of TA1 (0.01, 0.1, 1, and 10 μM) in the absence or presence of 5 μM KA or vehicle. In another set of experiments, slices were exposed to 10 μM T3, T1AM, TRIAC, T0AM, and TA0 in the absence or presence of 5 μM KA or vehicle.
Cell toxicity was evaluated 24 h later by measuring PI fluorescence (the fluorescence measured in KA-exposed slices in the CA3 region was reported as 100%).
Cell death in the presence of pharmacological modulation of PI3K/AKT and GSK-3β activities
Slices were preincubated for 5 min with vehicle or 10 μM LY294002 (a PI3K inhibitor; LY, Tocris) or 10 nM of the GSK-3β inhibitor TC-G (10 nM; Tocris) before adding TA1 (10 μM) or vehicle. All slices were then exposed to 5 μM KA, and cell death was evaluated 24 h later by measuring PI fluorescence.
Cell death in the presence of PYR
Slices were preincubated for 5 min with vehicle or 1 μM PYR, followed by TA1 (10 μM). All slices were then exposed to 5 μM KA. Cell death was assessed 24 h later by evaluating PI fluorescence.
Fluorescence was detected 30 min after the addition of PI using an inverted fluorescence microscope (Olympus IX-50; Solent Scientific) equipped with a xenon-arc lamp, a low-power objective (4 × ), and a rhodamine filter. Images obtained using a CCD camera (Diagnostic Instruments, Inc.) were digitized using the accompanying software (InCyt Im1™; Intracellular Imaging, Inc.) and subsequently analyzed using Image-Pro Plus morphometric analysis software (Media Cybernetics). To quantify cell death, the CA3 hippocampal subfield was identified and encompassed in a frame using the drawing function in ImageJ (National Institutes of Health), and PI fluorescence was then evaluated in arbitrary units (AU). Morphological analysis revealed a linear correlation between CA3 PI fluorescence and the number of injured CA3 pyramidal cells.
Preparation of acute rat hippocampal slices and whole-cell recordings
Brains were removed from Wistar rat pups and mounted in the slicing chamber of a vibroslicer (Leica VT 1000S). Horizontal slices (250 μm thick) were cut from the hippocampi in chilled artificial cerebral spinal fluid (aCSF) composed of 130 mM NaCl, 3.5 mM KCl, 1.25 mM NaH2PO4, 25 mM NaHCO3, 10 mM glucose, 2 mM CaCl2, and 1 mM MgSO4, and saturated with a 95% O2 + 5% CO2 gas mixture. Hippocampal slices were allowed to recover in the same solution maintained at 34°C with constant oxygenation for 1 h prior to the experiments. Slices were continuously perfused with the aCSF solution using a gravity-fed perfusion system. Whole-cell pipettes were filled with the following: 120 mM K+ gluconate, 15 mM KCl, 10 mM HEPES, 5 mM EGTA, 2 mM MgCl2, 5 mM Na2phosphocreatine, 0.3 mM Na2GTP, and 2 mM MgATP, resulting in a bath resistance of 3–5 MΩ.
Voltage-clamp experiments were performed using a PC-505B amplifier (Warner), and the results were digitized using Digidata 1440 A and Clampex 10 (Axon). Pipettes were pulled from borosilicate capillaries (Harvard Apparatus) using a Narishige PP830 vertical puller (Narishige International Ltd.). Pipette capacitance transients were cancelled, and no whole-cell compensation was utilized. Cells were maintained at −60 mV, and signals were sampled at 10 kHz and low-pass filtered at 1 kHz. All recordings were performed at 21–23°C.
KA (5 μM) was puff-applied for 10 s using a picospritzer (15). The combination of KA concentration and puff duration generated a non-desensitizing inward current. The KA puff was applied twice to exclude desensitization. Then, 10 μM TA1 was bath perfused for 5 min. At the end of TA1 application, KA was puff-applied once again.
Peak amplitude (pA) and peak area (nA*s) were calculated using Clampfit software.
Western blot analysis
Organotypic hippocampal slices treated as previously described were washed with cold 0.01 M phosphate-buffered saline (PBS; pH 7.4), blotted dry, and stored at −80°C until used in Western blot analysis.
Samples were gently transferred and dissolved in a lysis buffer tube containing 50 mM Tris HCl (pH 8), 150 mM NaCl, 1 mM EDTA, 0.1% w/v sodium dodecyl sulfate, and a protease and phosphatase inhibitor cocktail (Thermo Fisher Scientific). Total protein levels were quantified using the Pierce Protein Assay/bicinchoninic acid.
Proteins (20 μg) isolated from hippocampal slices or from mouse hippocampi were separated via 4–20% sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred into polyvinylidene fluoride membranes (60 min at 398 mA) using standard procedures. Blots were incubated overnight at 4°C with specific antibodies against p-AKT S473 (AB_329825), p-GSK3β S9 (AB_2115201), p-mTOR S2448 (AB_330970), phospho-p70S6K T389 (AB_330944; Cell Signaling Technology), c-fos (AB_259739; Sigma–Aldrich SRL), and α-tubuline (AB_310035; Merck-Millipore). Primary antibodies were diluted in PBS containing 1% albumin or 5% nonfat dry milk and 0.05% Tween. The antigen–antibody complexes were visualized using appropriate secondary antibodies (1:10,000, diluted in PBS containing 1% albumin or 5% nonfat dry milk and 0.05% Tween) and incubated for 1 h at room temperature. Blots were then extensively washed with PBS containing 0.1% Tween and developed using an enhanced chemiluminescence detection system (Pierce). Exposition and developing time were standardized for all blots. Densitometric analysis of scanned images was performed on a Macintosh iMac computer using the public domain NIH Image program. Results are presented as the mean ± standard error of the mean (SEM) of different gels and expressed as AU, which depict the ratio between levels of target protein expression and α-tubuline normalized to basal levels.
Statistical analysis
Statistical analyses consisted of one- or two-way analyses of variance (ANOVA) followed by post hoc tests (Bonferroni test and Tukey test for two- and one-way ANOVA tests, respectively) or unpaired t-test. When the experimental setting included only two groups, unpaired t-tests were used. Analyses of seizure occurrence were performed using chi-square tests with Yates correction. The threshold of statistical significance was set at p < 0.05. Data analysis was performed using GraphPad Prism v5.0 (GraphPad Software, Inc.). Results are expressed as the mean ± SEM.
Results
TA1 protects mice from PTZ-induced convulsions
The potential efficacy of TA1 in reducing neuron hyper-excitability was examined in a model of drug-induced convulsions (i.e., single administration of 90 mg/kg PTZ). The results indicate that 90 mg/kg PTZ s.c. induced convulsions in 100% of mice, with a mean latency to first seizure of 548 ± 38.82 s. The chi-square test with Yates correction of results indicated that the treatment with 7 and 11 μg/kg TA1 significantly reduced the number of mice experiencing convulsions (p = 0.0183 and p = 0.0047 vs. vehicle, respectively; Fig. 1A). Furthermore, the ANOVA test and the post hoc analysis indicated that 7 and 11 μg/kg TA1 significantly increased (p < 0.01 and p < 0.05 vs. PTZ, respectively) the latency to first seizure (Fig. 1B) with respect to PTZ plus vehicle-treated mice.
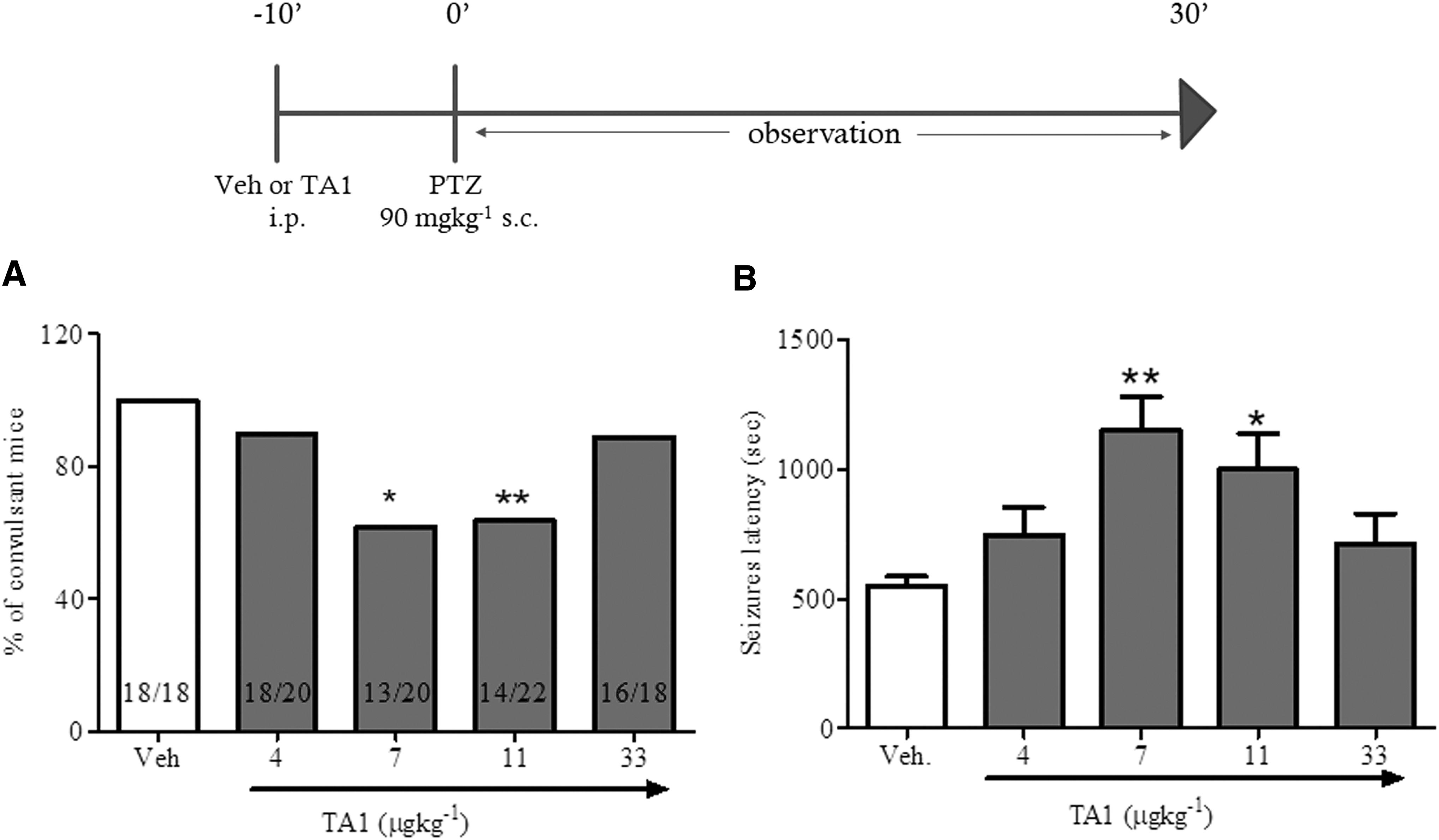
3-Iodothyroacetic acid (TA1) decreases the percentage of convulsant mice and increases seizure latency. Mice pretreated with TA1 (4, 7, 11, and 33 μg/kg, i.p.) or vehicle (Veh.) were treated 10 min later with pentylenetetrazole (PTZ; 90 mg/kg). Mice were observed for 30 min, and the number of mice presenting convulsions and the latency to first clonic seizure occurrence were recorded. (
TA1 activates hippocampal PI3K/AKT pathway and increases the phosphorylation levels of GSK-3β
Next, the study evaluated whether TA1 is able to activate the PI3K/AKT cascade. Mice treated with 7 μg/kg TA1 i.p. or with vehicle were sacrificed 24 h later to collect hippocampi for Western blotting analysis.
The results show that in mice treated with 7 μg/kg TA1, hippocampal levels of p-AKT, p-GSK-3β (p = 0.0491 and p = 0.0157, respectively; unpaired t-test) and also of p-mTOR (p = 0.0012; unpaired t-test; Fig. 2A and B) were significantly higher than in vehicle-treated mice. However, no changes in the phosphorylation levels of p70S6K, the downstream effector of the mTORC1 complex, were observed (data not shown).
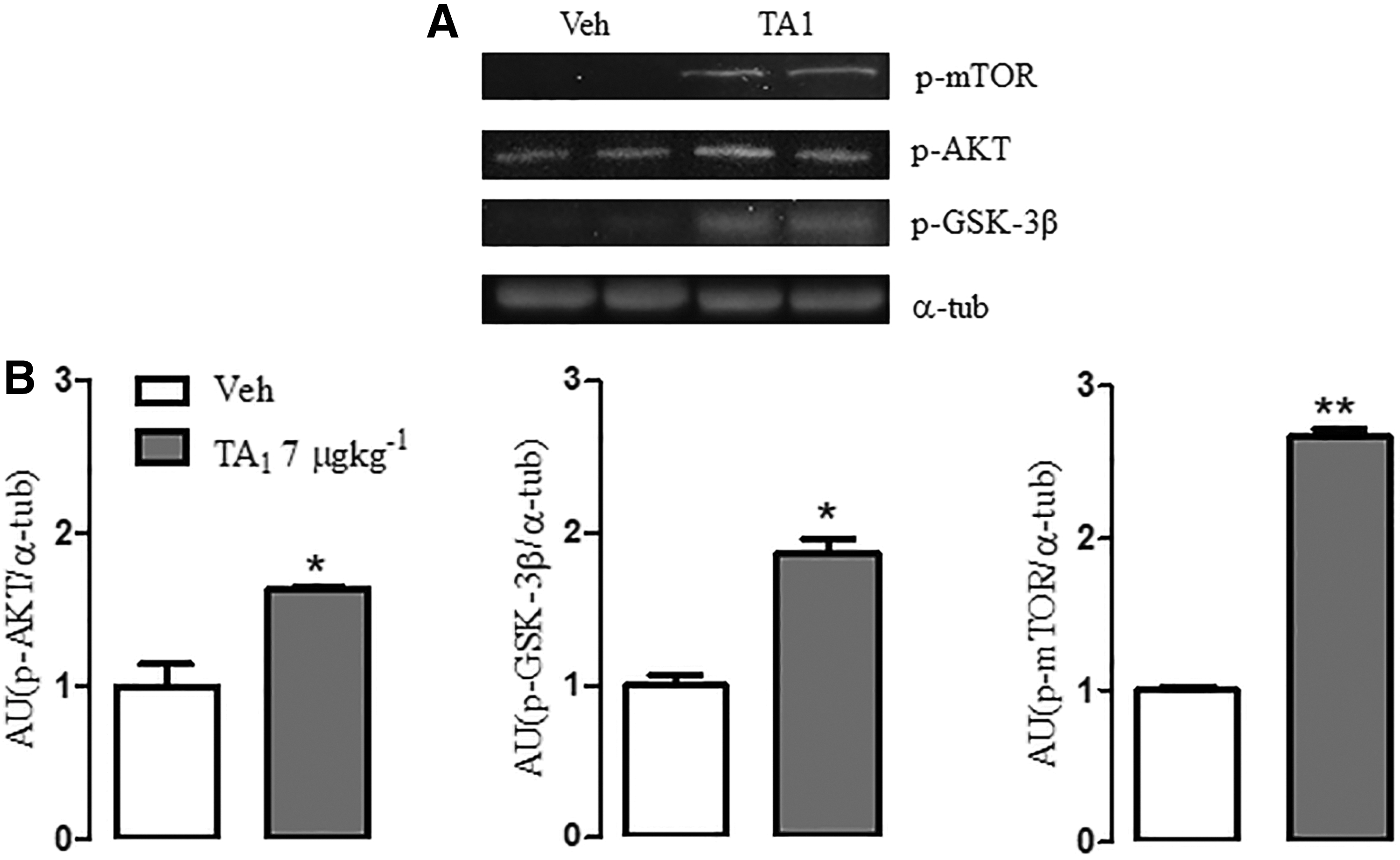
TA1 activates the PI3K/AKT pathway and increases phosphorylation levels of the GSK-3β in mouse hippocampus. Male CD1 mice received an intraperitoneal injection of either vehicle (Veh) or 7 μg/kg TA1. Hippocampi were isolated from mice sacrificed 24 h following treatment. Levels of p-AKT, p-GSK-3β, and p-mTOR were assessed by Western blotting. (
TA1 restores hippocampal c-fos levels reduced by PTZ
It was reported that PTZ reduces hippocampal levels of c-fos (15,16), an early gene product that is part of the pro-survival PI3K/AKT cascade.
Accordingly, c-fos levels were measured in the hippocampi of mice treated i.p. with vehicle, PTZ, or with PTZ plus 7 μg/kg TA1 (PTZ + TA1). One-way ANOVA confirmed that PTZ treatment significantly reduces hippocampal c-fos levels compared to vehicle-injected mice (p < 0.001). Interestingly, c-fos levels in the hippocampus of mice that had received PTZ + TA1 were similar to levels measured in vehicle-treated mice (p > 0.05; Fig. 3A and B).
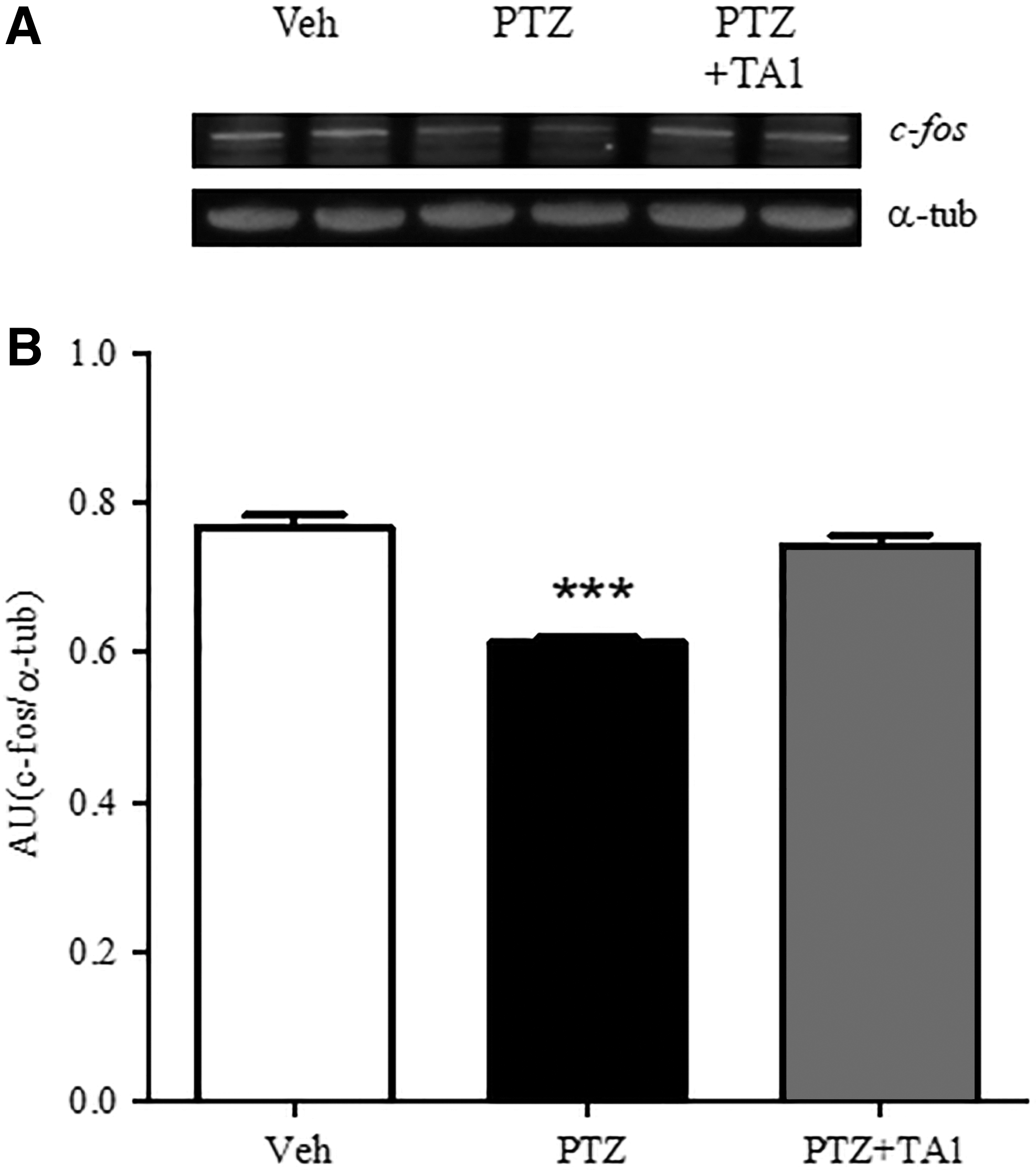
TA1 restores hippocampal c-fos levels reduced by PTZ. Hippocampi were isolated from mice treated 24 h before with vehicle (Veh; n = 3), PTZ (90 mg/kg; n = 3), or PTZ plus 7 μg/kg TA1 (PTZ + TA1; n = 3). c-fos expression levels were analyzed by Western blotting. c-fos levels were significantly reduced by PTZ treatment. Results from a representative experiment (
TA1 does not induce cytotoxicity in organotypic hippocampal slices
Organotypic hippocampal slices are currently considered a model to study excitotoxic neuronal death (12 –14) and the associated signaling. Organotypic hippocampal slices exposed to vehicle or increasing concentrations of TA1 (0.01, 0.1, 1, and 10 μM) for 24 h exhibited similar low levels of PI fluorescence, indicating that TA1 did not induce cell toxicity at any of the tested concentrations.
KA-induced toxicity in the CA3 region: the effect of TA1, T3, and other T3 metabolites
To mimic excitotoxicity, slices were exposed to vehicle or to 5 μM KA for 24 h. Under such conditions, selective cell death induced by KA in the CA3 region (PI fluorescence in CA3: 100%) was noted. The exposure of slices to TA1 (0.01, 0.1, 1, 10 μM) and then to 5 μM KA showed a lower cell death rate in the CA3 region than in slices treated only with KA (5 μM). The extent of cell death became significant in slices exposed to 10 μM TA1 (p < 0.001; Fig. 4A and B).
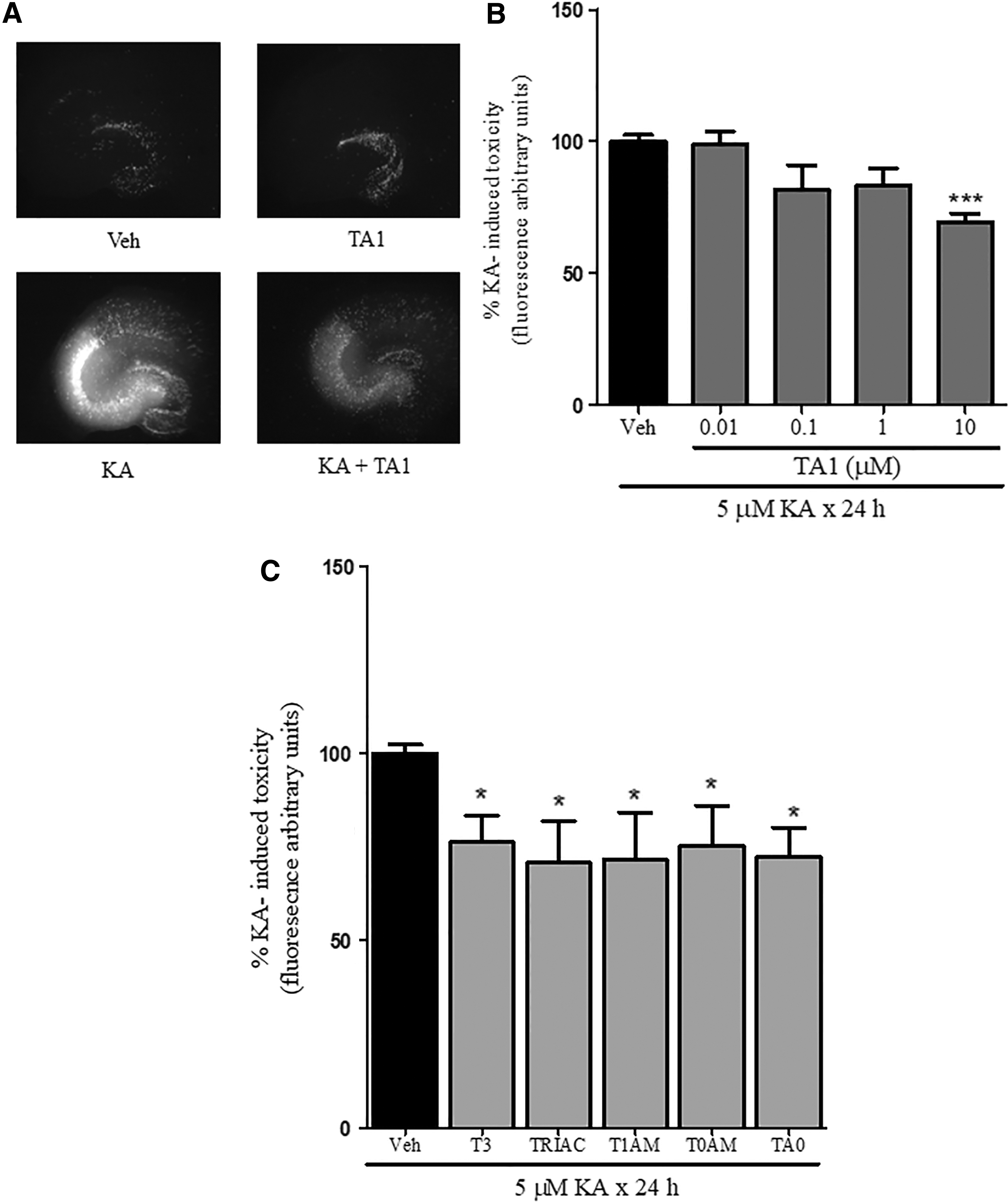
TA1 and other T3 metabolites reduce kainic acid (KA)-induced neuron death. Organotypic hippocampal slices were exposed to vehicle (Veh) or to 5 μM KA for 24 h in the absence (Veh) or presence of 0.01, 0.1, 1, or 10 μM TA1. Cell death was evaluated by measuring propidium iodide (PI) levels of fluorescence 24 h after treatment. Fluorescence levels measured in hippocampal slices exposed to KA were associated with neuronal death in the CA3 region. Drug effect was reported as a percentage of KA toxicity value (regarded as 100%). (
To verify whether neuroprotective activity was conserved by other T3 metabolites with different iodide content, slices were exposed to 10 μM T3, T1AM, TRIAC, T0AM, and TA0. The results indicate that all the compounds tested reduce KA-induced neuron cell death in the CA3 region (Fig. 4C), thus suggesting that neuroprotection is likely a common feature of T3 metabolites.
TA1 activates the PI3K/AKT cascade in hippocampal slices
Next, the study examined whether TA1 is also able to activate the PI3K/AKT and to increase GSK-3β phosphorylation in organotypic hippocampal slices. Slices were exposed to 10 μM TA1, a concentration found to reduce KA-induced cell death, and the phosphorylation levels of these kinases were measured. The results indicate that the exposure of slices to 10 μM TA1 for 24 h resulted in a significant increase in p-AKT and p-GSK-3β levels relative to that observed in vehicle-treated slices (p = 0.0016 and p = 0.0098 for p-AKT and p-GSK-3β, respectively; unpaired t-test; Fig. 5A and B). As in the mouse model, the activation of this cascade was associated with increased levels of p-mTOR but not of phospho-p70S6K (p < 0.0001 vs. vehicle; unpaired t-test; Fig. 5A and B; data not shown).
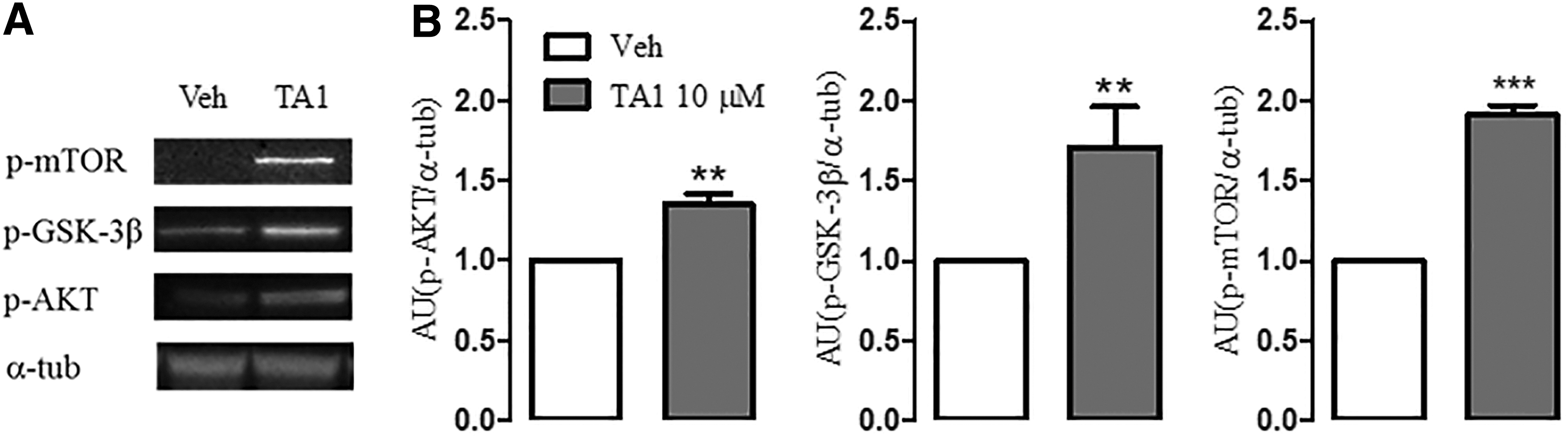
TA1 activates the IP3/AKT cascade in organotypic hippocampal slices. Slices were exposed to Veh or to 10 μM TA1 for 24 h, following which levels of p-AKT, p-GSK-3β, and p-mTOR were evaluated. (
Other T3 metabolites activate AKT
Then, the study examined whether the other T3 metabolites studied activate the AKT pathway. The results demonstrate that pAKT levels were higher in slices exposed to 10 μM T3, TRIAC, T1AM, T0AM, and TA0 for 24 h than in slices exposed to vehicle (Fig. 6A and B).
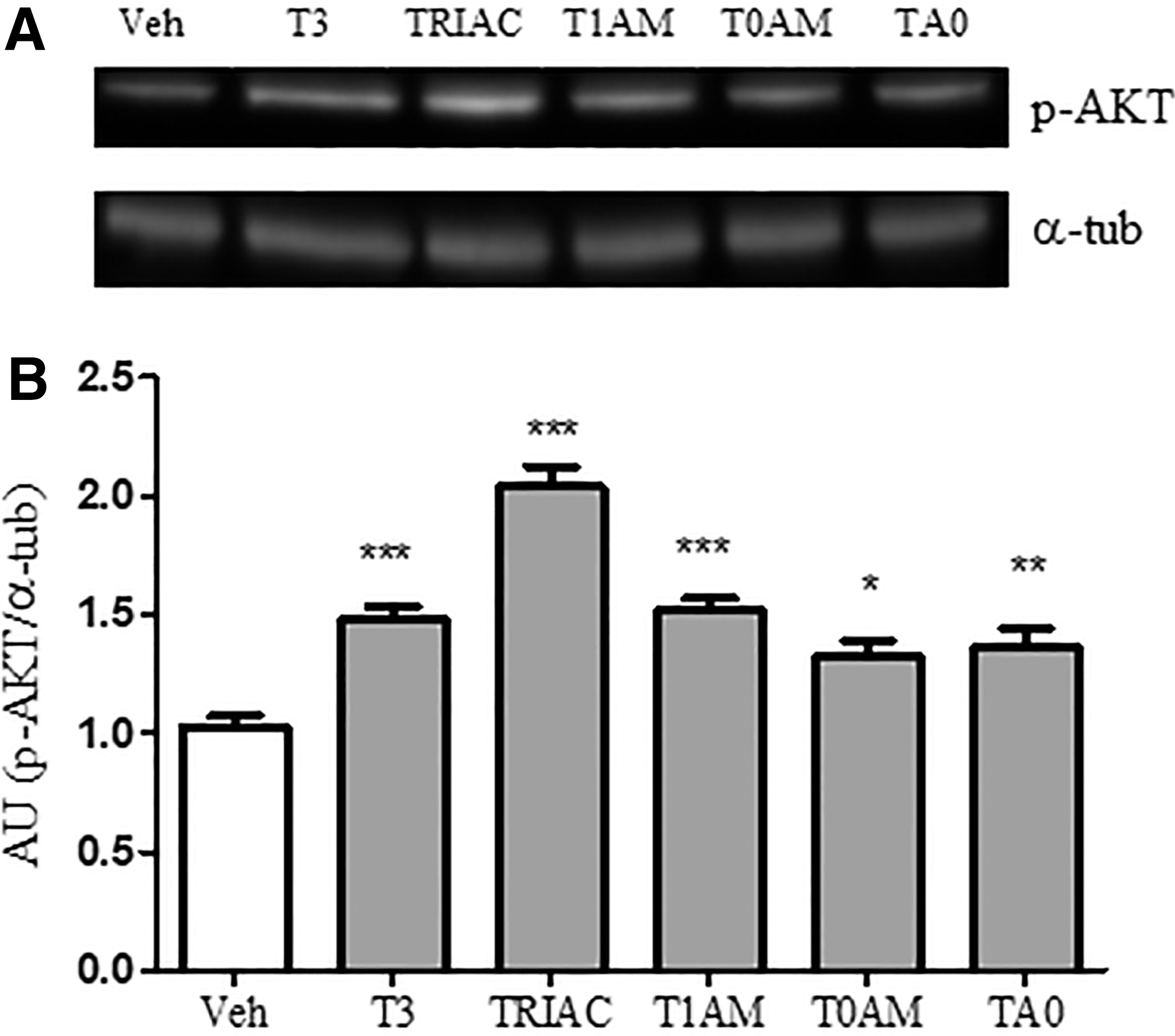
Other T3 metabolites increase p-AKT hippocampal levels. Slices were exposed to Veh or to 10 μM T3, TRIAC, T1AM, T0AM, or T0A for 24 h. p-AKT expression was then determined. (
TA1-induced reduction of neuronal death depends on PI3K/AKT and on GSK-3β activity
The study investigated whether activation of PI3K/AKT and inhibition of GSK-3β are involved in TA1-induced reduction of KA-induced cell death. Slices were pretreated with vehicle or 10 μM LY294002 or 10 nM TG-C, inhibitors of AKT and of GSK-3β, respectively, before adding TA1 (10 μM) and KA (5 μM).
The results indicate that: (i) 10 μM LY294002 does not modify KA-induced neuronal toxicity (100%) but prevents the neuroprotective effects of 10 μM TA1; (ii) 10 nM TC-G significantly reduces KA toxicity to 72.67 ± 4.2% (p < 0.05; Fig. 7A and B); and (iii) in the presence of 10 nM TC-G and of 10 μM TA1, KA-induced cell death is reduced further (35.24 ± 9.62%; Fig. 7C and D).
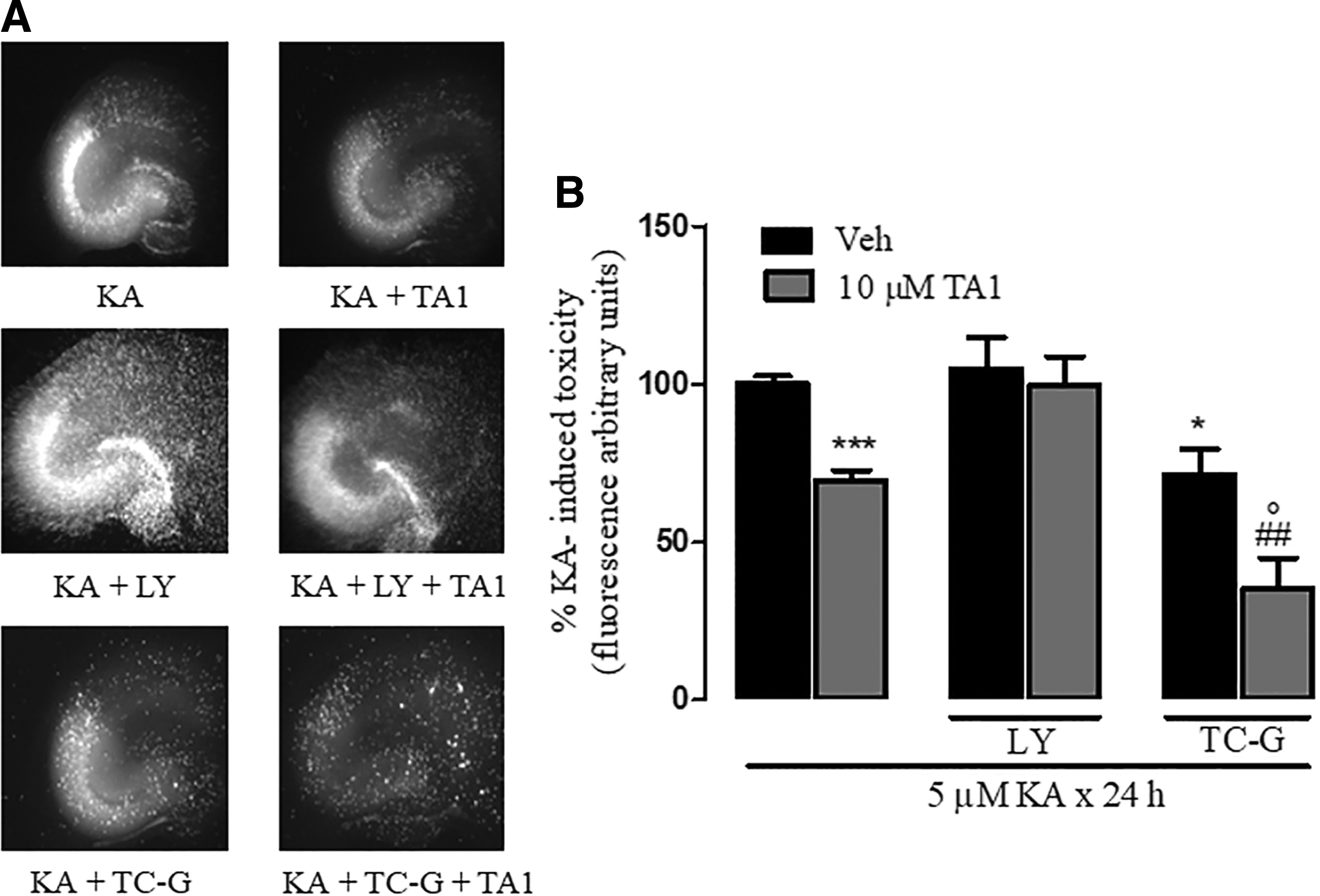
TA1-induced reduction of neuronal death depends on the activation of PI3K/AKT and GSK-3β. (
Anticonvulsant and neuroprotective effects of TA1 are prevented by PYR
To investigate for a possible histamine involvement, 7 μg/kg TA1 or vehicle was administered to mice pretreated with PYR (10 mg/kg i.p.) or vehicle (i.p.), and then they all received PTZ (90 mg/kg), as described in the Methods. As expected, in mice pretreated with vehicle, 7 μg/kg TA1 significantly increases seizure latency (p < 0.001; vs. PTZ) and reduces the percentage of mice presenting with convulsions (p = 0.0362 vs. vehicle; chi-square test with Yates correction; Fig. 8A and B). It is worth noting that PYR treatment abolished the protection offered by TA1 either on the number of convulsing mice or on the latency of first seizure onset.
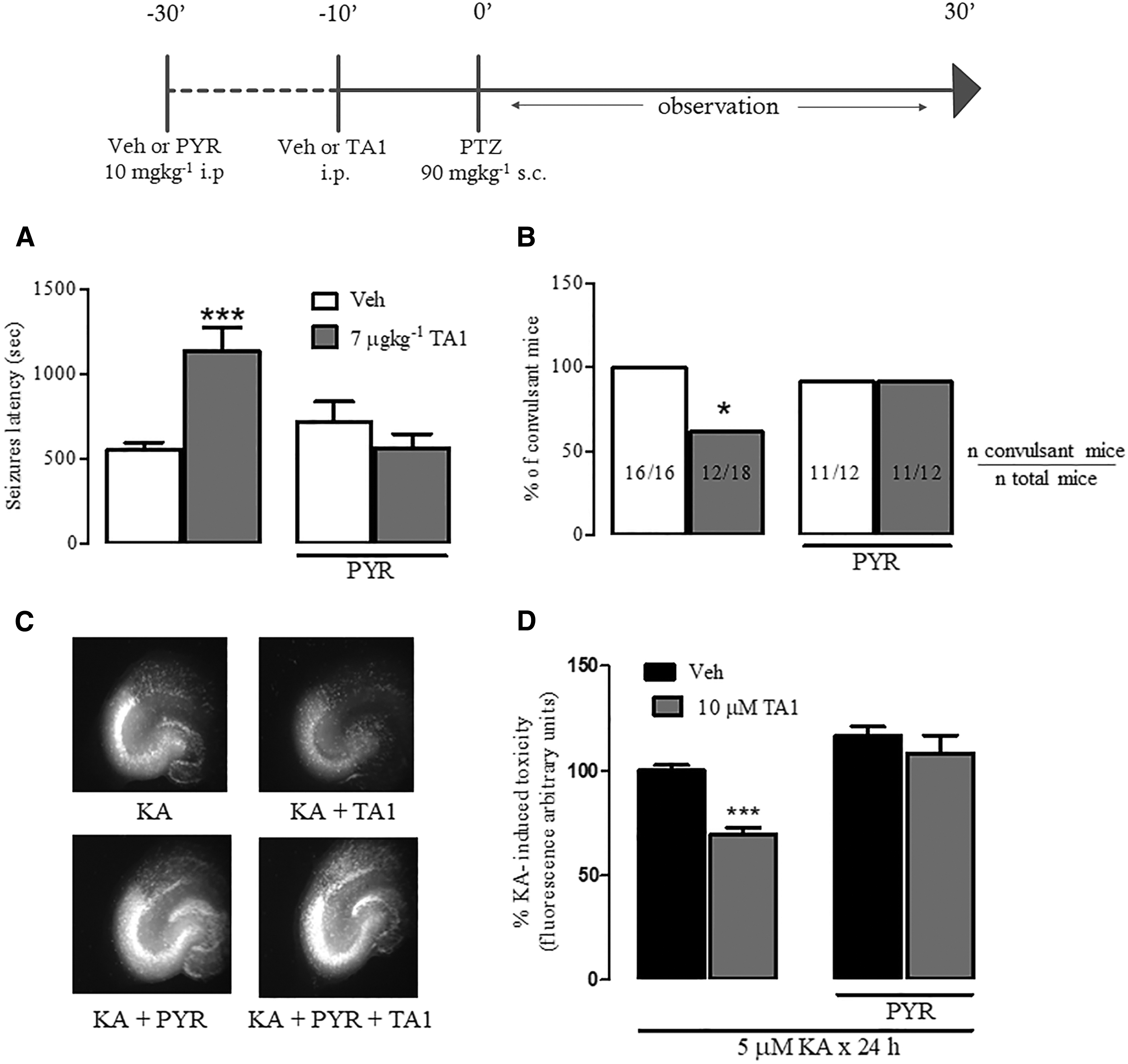
Anticonvulsant and neuroprotective effects of TA1 are prevented by pyrilamine (PYR). Mice were pretreated with 10 mg/kg PYR, after which they received vehicle (Veh) or 7 μg/kg TA1 prior to treatment with PTZ (90 mg/kg). (
The effect of PYR (1 μM) on TA1-induced protection against KA-induced cell death was then evaluated in organotypic hippocampal slices. The ANOVA test of results indicates that there was a significant effect of pretreatment PYR (p < 0.001) and of TA1 treatment (10 μM; p < 0.001), and a significant interaction of between pretreatment and treatment (PYR + TA1; p < 0.05) in reducing KA toxicity. Post hoc testing showed that PYR itself exerts no effect on KA-induced toxicity, but its presence completely prevented the neuroprotective effects of TA1 (p > 0.05 vs. vehicle). Consistently, the levels of PI fluorescence in PYR + TA1 + KA slices were not significantly different from those observed in slices exposed to KA alone (100%; Fig. 8C and D).
TA1 does not interfere with KA-induced inward currents in acute brain slices
The effects of TA1 on ionotropic glutamate receptors were investigated by measuring KA-mediated currents in CA3 pyramidal neurons following treatment with TA1. The results indicate that TA1 does not significantly modify the peak amplitude of KA-induced inward currents or peak area (Fig. 9A and B).

TA1 does not interfere with KA-induced inward currents in acute brain slices. KA-induced currents were evaluated via voltage clamping of neurons from the CA3 region of acute hippocampal slices. KA (5 μM) was puff-applied for 10 s using a picospritzer. (
Discussion
The findings indicate that TA1, a thyroid hormone metabolite, protects against PTZ-induced neuron hyper-reactivity, acting as an anticonvulsant, and that it activates pathways involved in protecting neurons from excitotoxic insults. The anticonvulsant and neuroprotective effects of TA1 were prevented by pretreating mice with PYR, and were independent of TA1 modulation on KA receptors. Interestingly, T3 and TETRAC, T1AM, T0AM, and TA0 shared the capacity to protect hippocampal neurons from excitotoxicity with TA1.
Neuron hyper-excitability was induced in mice by administering PTZ, a selective inhibitor of the GABAergic transmission (11). As a result, mice receiving PTZ were prone to seizures. Dong et al. (16) reported that PTZ-induced hyper-excitability can produce neurotoxic effects, since this treatment is associated with reduction of c-fos hippocampal levels. In fact, c-fos is an early gene product that controls the expression of neuroprotective factors including BDNF. This latter peptide is in turn a target of AKT and GSK-3β pathways (17,18).
Under the present experimental conditions, PTZ induced convulsions in 100% of subjects. This number was significantly reduced when mice received 7 and 11 μg/kg TA1. In addition, 7 and 11 μg/kg TA1 also increased the latency to first seizure. The anticonvulsant effect of TA1 showed an inverted U-shaped dose–effect relationship, thus suggesting a rapid desensitization (19). In agreement with previous findings, it is now confirmed that treatment with PTZ is associated with the reduction of c-fos hippocampal levels. Interestingly, the reduction of c-fos expression was prevented by an effective anticonvulsant dose of TA1. Overall, TA1 acutely counteracts the effects of PTZ on neuronal hyper-excitability and c-fos transcription levels, thus suggesting a neuroprotective potential of the acid. The data obtained in organotypic hippocampal slices confirm that TA1 is indeed neuroprotective against KA-induced cell death.
In fact, the results from organotypic hippocampal slices indicate that TA1 reduces KA-induced cell death in the CA3 region, the region that is known to contain histaminergic neurons (20). The results also indicate that this kind of neuroprotection is also mediated by T3 and other T3 metabolites, although it remains undetermined whether these metabolites have anticonvulsant activity. Of note, effective TA1 concentrations in vitro are higher than those effective in vivo. This is due to the well established poor diffusion of drugs across organotypic slices (12 –14). In this respect, TA1 shows a trend to protection at 0.1 μM achieving statistical significance at 10 μM.
The mechanism of TA1-mediated protection was explored, and it was found that the acid has the intrinsic capacity to activate a molecular “protective ground,” which buffers the impact of possible negative insults (hyper-excitability or excitotoxicity) on neuron survival. In fact, it is demonstrated that TA1 treatment of mice increase levels of pAKT and of GSK-3β (kinase inhibition) in the hippocampus. This signaling pathway cascade was also found to be activated in organotypic hippocampal slices exposed to TA1 for 24 h. Furthermore, based on the in vitro studies, it can be concluded that inhibition of AKT and of GSK-3β phosphorylation are necessary for TA1-induced neuroprotection, as TA1 neuroprotection was lost in slices pretreated with LY294002, an inhibitor of PI3K. Consistently, the protective effect of TG-C, an inhibitor of GSK-3β, was further increased in the presence of TA1.
The PI3/AKT/GSK-3β cascade is reported as a fingerprint of the non-genomic neuroprotective effects of T3 (2 –4). In line with this evidence, it is demonstrated that T3 and other T3 metabolites activate AKT in organotypic hippocampal slices. Furthermore, like T3, TA1 signaling activity in the hippocampus includes the activation of mTOR but, unlike T3, not that of phosho-p70S6K, the downstream effector of mTORC1 (4). Although the significance of mTOR activation by TA1 remains largely unknown, the absence of the phosho-p70S6K might indicate the involvement of TORC2 rather than TORC1 complexes. Overall, the evidence indicates that TA1-triggered signaling is maintained after its anticonvulsant action has dissipated.
Overall, based on the in vivo and in vitro findings, it is concluded that TA1, directly or indirectly, not only activates pathways “preparing” neurons to fight against excitotoxicity, but also that histamine is part of the neuroprotective program orchestrated by TA1. In fact, the dose-specific anticonvulsant and neuroprotective effect of TA1 against PTZ in vivo and KA in vitro were prevented by the H1 antagonist PYR, which was chosen based on past experience. In fact, although it shows some affinity for cholinergic as well as non-H1 histamine receptors, PYR is one among few H1R antagonists showing central bioavailability following systemic administration. Non-histaminergic effects of PYR are excluded, since it has already been demonstrated that at a dose of 10 mg/kg, PYR does not induce sedation, analgesia, or amnesia in mice (8,9,19). Furthermore, it has previously been demonstrated that TA1 does not interact with muscarinic receptors (21).
Conclusions
TA1 administration to mice exerts an immediate effect in reducing neuronal hyper-excitability (i.e., an anticonvulsant effect) and initiates a protective, pro-survival, signaling cascade that is maintained for 24 h after administration. Such activation also occurs in hippocampal preparations, and it is the mechanism, likely shared by other T3 metabolites, by which TA1 reduces neuron death by excitotoxicity. These findings expand the pharmacological profile of TA1, and suggest that TA1 could play a role as a novel anticonvulsant agent that also attenuates the detrimental effects of excitotoxicity on neuron survival.
Footnotes
Acknowledgments
This work was supported by a grant from University of Florence and from Ente Cassa di Risparmio di Firenze (2017).
Author Disclosure Statement
The authors declare no conflict of interest.