
Abstract
Background:
Thyroid cancer is an emerging health problem in the United States and worldwide. With incidence rates of thyroid cancer rapidly rising, the need to develop new treatment options is becoming a priority, and understanding the molecular mechanisms of this disease is crucial to furthering these efforts. Thyroid growth is driven by the TSH/cAMP/PKA signaling pathway, and it has previously been shown that activation of PKA through genetic ablation of the regulatory subunit Prkar1a (Prkar1a KO) is sufficient to cause follicular thyroid cancer in mouse models. cAMP also activates the Epac proteins and their downstream effectors, Rap1a and Rap1b.
Methods:
Previously, the authors' laboratory generated a mouse model of follicular thyroid cancer by conferring thyroid-specific deletion of Prkar1a (R1a-TpoKO). To probe the roles of other components of the PKA signaling system in the development of thyroid cancer, this study deleted Rap1 and Epac1 in the setting of the Prkar1a knockout.
Results:
Deletion of Rap1 significantly decreases thyroid size and cancer incidence in Prkar1a KO thyroids. Further, isoform-specific ablation of Rap1a and Rap1b implicates Rap1b as the downstream effector of PKA during thyroid carcinogenesis. In vivo modeling provides definitive evidence that Epac1 plays little role in thyroid proliferation and is dispensable for thyroid carcinogenesis arising from the deletion of Prkar1a.
Conclusions:
This study demonstrate that PKA signaling to Rap1b is a key signaling node for follicular thyroid carcinogenesis, while Epac1 activity is not required for tumor development. This work sheds new light on the pathways involved in FTC development and identifies a possible target for the development of new therapies in the treatment of FTC.
Introduction
T
In humans, loss-of-function mutations in PRKAR1A, which encodes the type 1a regulatory subunit of the cAMP dependent protein kinase A (PKA), cause the inherited tumor predisposition syndrome Carney Complex (3). Carney Complex is characterized by tumors in multiple tissues, including FTC, as a part of the disease spectrum (3,4). In mice, thyroid-specific deletion of Prkar1a (Tpo-R1aKO) leads to locally invasive thyroid carcinoma as a result of over-activation of the PKA pathway (5). This mouse model of FTC serves as a useful tool in the study of PKA-mediated thyroid carcinogenesis, as previous mouse models of increased PKA signaling through adenylyl cyclase have not developed cancer (6 –9).
In the thyroid, PKA is known to signal downstream of thyrotropin (TSH) through the production of cAMP, and increased levels of TSH are associated with thyroid carcinogenesis in humans (10,11). As such, cAMP has been a popular object of research for targeted drug therapies. However, given the wide range of physiological responses regulated by cAMP, pharmacological manipulation of cAMP levels can have an array of side effects, including emesis and cardiac dysfunction (12). In an attempt to circumvent this problem, recent research has focused on downstream targets of cAMP for a more targeted approach to cAMP pathway manipulation. Rap1 is a small GTPase containing two isoforms, each encoded by a separate gene, Rap1a and Rap1b. Rap1 activity has been shown to be regulated by both cAMP and PKA through signaling by TSH (13). Increased Rap activity is associated with many cancers, including thyroid cancer, and it has been suggested that Rap1 dysregulation can contribute to malignancy (9,14 –17).
Exchange protein directly activated by cAMP (Epac) is another intracellular receptor that serves to mediate the effects of cAMP to activate the common PKA/Epac target Rap1 (18,19). Epac has two isoforms: Epac1 and Epac2. Epac1 is ubiquitously expressed, with particularly high expression in tissues such as the kidney, ovary, and thyroid, while Epac2 is selectively expressed in a limited number of tissues, with no detectable expression in the thyroid (19,20). Epac has been shown to regulate Rap activity both in concert with PKA and independently, and the effects—whether stimulatory or inhibitory—seem to be dependent on cellular context and the type of stimuli (13,18,21,22). Further, Epac1 has been shown to play a role in cell migration and invasion in other cancers (21,23). Thus far, discriminating between PKA- and Epac-dependent Rap1 activation in vitro has proved difficult, as they have similar affinities for many cAMP analogs, although analogs with selective binding properties have been developed. However, these analogs are not usable in vivo. Therefore, in vivo gene- and isoform-specific knockout systems are needed to study the detailed interactions among these proteins further.
This study used murine knockout models to probe the roles of Rap1 and Epac1 in Prkar1a-associated thyroid carcinogenesis. The study demonstrates that loss of Rap1 in Prkar1a KO thyroids significantly reduces the risk of developing thyroid cancer, highlighting a central role of PKA-Rap1 signaling in the development of FTC. Further, it shows that Epac1 does not appear to play a significant role in mediating cAMP-PKA-Rap1 signaling in thyroid tumorigenesis. These studies further our understanding of the molecular mechanisms of FTC progression and may help direct future research into the development of novel therapeutics.
Methods
Animal studies
TPO-cre;Prkar1aloxP/loxP mice (5) were bred with Rap1aloxP/loxP;Rap1bloxP/loxP animals (24) to generate thyroid-specific deletions of total Rap1 and individual Rap1 isoforms. For studies involving Epac1, TPO-cre;Prkar1aloxP/loxP mice were crossed with global Epac1–/– (25) mice.
Three-dimensional thyroid ultrasonography was performed at three-month intervals on live animals using a VisualSonics Vevo2100 Ultrasound equipped with an MS550D transducer. Images were acquired in 3D mode and volumes calculated using Vevolab v2.1.
Goitrogen treatment was given to Epac–/– and wild-type (WT) littermate mice for six months. Treatment consisted of methimazole (0.5 g/L) and sodium perchlorate (5 g/L) in drinking water ad libitum changed twice weekly (9). Mice were also maintained on a low-iodine diet (Envigo Teklad TD.120460).
Histology
Dissected mouse thyroid tissues were fixed in 10% buffered formalin solution and embedded in paraffin. Tissue sections were cut in 5 μm sections and stained with hematoxylin and eosin. Immunohistochemistry was performed, as previously described (26), with the following antibodies: Epac1 (ab21236; Abcam), Ki-67 (ab15580; Abcam), and cleaved Caspase-3 (9661; Cell Signaling Technology). Quantification of cleaved Caspase-3 staining was performed using ImageJ software to measure the area of positive staining divided by the total area in pixels. Ki-67 staining is represented as a ratio of number of Ki-67+ cells per field divided by total number of cells per field in 40 × images.
Western blotting
Proteins from mouse tissues were prepared and run on sodium dodecyl sulfate polyacrylamide gels, followed by transfer to nitrocellulose membrane. Membranes were blocked with 2% bovine serum albumin and probed with antibodies against Rap1 (07-916; Millipore) and β-actin (3700S; Cell Signaling Technology)
Thyroid hormone assay
Blood was collected from 10 animals per genotype and centrifuged for 20 min at 4°C to separate serum from whole blood. Serums were shipped on dry ice to Vanderbilt University Medical Center Hormone Assay and Analytical Services Core (part of the Mouse Metabolic Phenotyping Center) for thyroxine (T4) concentration determination using double antibody radioimmunoassay.
Statistics
All data, except cancer incidence rates, were analyzed via unpaired t-test using Prism GraphPad software. p-Values of <0.05 were considered significant. Tumor incidence rates were compared using a logistic regression and controlled for multiple comparisons using Holm's procedure. Incidence rates are represented as a percent incidence with confidence intervals.
Results
Thyroid specific deletion of Rap1 decreases incidence of Prkar1a-mediated FTC formation
This project began as an effort to assess the role of Rap1 in Prkar1a-mediated thyroid carcinogenesis. However, before addressing this point, it was first necessary to examine the effect of Rap1 KO on normal thyroid function. To this end, first a thyroid-specific deletion of Rap1 isoforms was generated by crossing thyroid peroxidase (TPO)-cre mice with mice harboring conditional null alleles of Rap1a and Rap1b (24) to obtain TPO-cre;Rap1aloxP/loxP
and TPO-cre;Rap1bloxP/loxP
animals, henceforth referred to as Tpo-Rap1A and Tpo-Rap1B. Conditional deletion of either Rap isoform in the thyroid had no effect on thyroid morphology or histology (Supplementary Fig. S1A and B; Supplementary Data are available online at
To determine if Rap1 plays a role in Prkar1a-mediated thyroid tumorigenesis, Tpo-Cre;Prkar1aloxP/loxP mice were crossed with Rap1aloxP/loxP;Rap1bloxP/loxP animals to generate Tpo-cre;Prkar1aloxP/loxP;Rap1aloxP/loxP;Rap1bloxP/loxP mice, henceforth referred to as Tpo-R1aRap1KO. Knockout was confirmed by Western blot (Fig. 1A), showing decreased Rap1 protein relative to Tpo-R1aKO mouse thyroids. As the Tpo-cre driver is highly efficient, residual Rap1 detected in the blots most likely arises from cells outside of the thyroid follicular lineage. Tpo-R1aRap1KO mice and WT littermates were observed for nine months (the time after which mice began dying), and subjected to ultrasonography at three-month intervals to record thyroid growth over time. At the study endpoint, Tpo-R1aRap1KO thyroids were significantly smaller than age-matched Tpo-R1aKO thyroids (20.584 ± 23.13 mm3 vs. 47.07 ± 24.72 mm3, respectively; p = 0.0011; Fig. 1B–E). Size differences between Tpo-R1aRap1KO and WT littermate controls showed a trend toward mild enlargement, which did not reach statistical significance (7.66 ± 1.84 mm3 for WT, p = 0.063, compared to Tpo-R1aRap1KO; Fig. 1D). No sex differences were found in the thyroid sizes of Tpo-R1aRap1KO mice (p = 0.945; data not shown). Phenotypically, this genetic model of Prkar1a-induced thyroid cancer exhibits complete involvement of the thyroid gland. As such, the entire thyroid is considered tumorous in Tpo-R1aKO mice (Fig. 1G). Histological analysis of Tpo-R1aRap1KO thyroids revealed that while many thyroids exhibited increased cellularity, the majority of Tpo-R1aRap1KO thyroids had regained substantial macrofollicular structures and lacked indicators of a carcinogenic phenotype, including capsular fibrosis and local invasion (Fig. 1F–H). Incidence rates of thyroid carcinoma in Tpo-R1aRap1KO were significantly lower than incidences in age-matched Tpo-R1aKO mice (27.78% incidence vs. 81.8%, respectively; p = 0.0091), indicating that deletion of Rap1 significantly reduces the risk of developing thyroid tumors in the setting of Prkar1a KO.

Thyroid-specific deletion of Rap1 in Tpo-R1aKO mice reduces thyroid size. (
Rap1b is the isoform that confers resistance to thyroid tumor formation in Tpo-R1aKO mice
To determine which isoform of Rap1 is responsible for the reduction in cancer incidence, single isoform knockouts were generated in the setting of Prkar1a deletion. Tpo-R1aRap1AKO (Tpo-Cre;Prkar1aloxP/loxP;Rap1aloxP/loxP ) had thyroid volumes similar to Tpo-R1aKO mice (61.24 ± 4.12 mm3, p = 0.35, compared to Tpo-R1aKO; Fig. 2A, C, and E). Due to decreased survival of the single isoform deleted Tpo-R1aRap1AKO mice (due to a nonthyroidal effect of the conditional allele), it was necessary to combine the six- and nine-month time point for phenotyping purposes, resulting in comparable incidence rates of carcinoma between Tpo-R1aRap1AKO mice at six to nine months of age compared to 9-month-old Tpo-R1aKO mice (80%; p = 0.9075 vs. Tpo-R1aKO). Conversely, selectively deleting the 1B isoform of Rap in Tpo-R1aRap1BKO thyroids resulted in a reduction in thyroid size relative to Tpo-R1aKO mice (18.93 ± 10.60 mm3 vs. 47.07 mm3, respectively; p = 0.0039; Fig. 2B, D, and F).

Decreases in thyroid size in Tpo-R1aRap1KO mice is mediated by Rap1b. (
At the histologic level, the range of features of Tpo-R1aRap1AKO thyroids was comparable to that of Tpo-R1aRap1BKO thyroids (Fig. 3A and B), although Tpo-R1aRap1KO and Tpo-R1aRap1BKO mice exhibited a clear decrease in carcinoma incidence relative to Tpo-R1aRap1A mice(44% carcinomas for Tpo-R1aRap1BKO vs. 80% carcinomas for Tpo-R1aRap1AKO; p = 0.044; Fig. 3C), indicating that Rap1b is the predominant isoform required for Prkar1a-mediated tumorigenesis. To understand better the role Rap1b plays in reduction of tumor size, expression of apoptosis and proliferation markers were examined. While proliferation was unchanged between models (4.03 ± 2.55 Ki-67+ cells Tpo-R1aRap1A vs. 3.78 ± 1.38 Ki-67+ cells Tpo-R1aRap1B; p = 0.79; data not shown), Tpo-R1aRap1B thyroids expressed significantly higher levels of cleaved Caspase 3 relative to Tpo-R1aRap1AKO tumors (Fig. 3F, quantified in Fig. 3D), implicating an increase in cell death as the driving force behind the lack of excessive tumor growth in Tpo-R1aRap1KO or Tpo-R1aRap1BKO thyroids. In fact, caspase staining tended to involve entire follicles, which exhibited a solid appearance with caspase staining throughout.

Tpo-R1aRap1BKO mice have a lower incidence rate of thyroid carcinoma due to increases in apoptosis relative to Tpo-R1aRap1A mice. (
Epac1 is not a major component in the PKA-Rap1 thyroid tumorigenic cascade
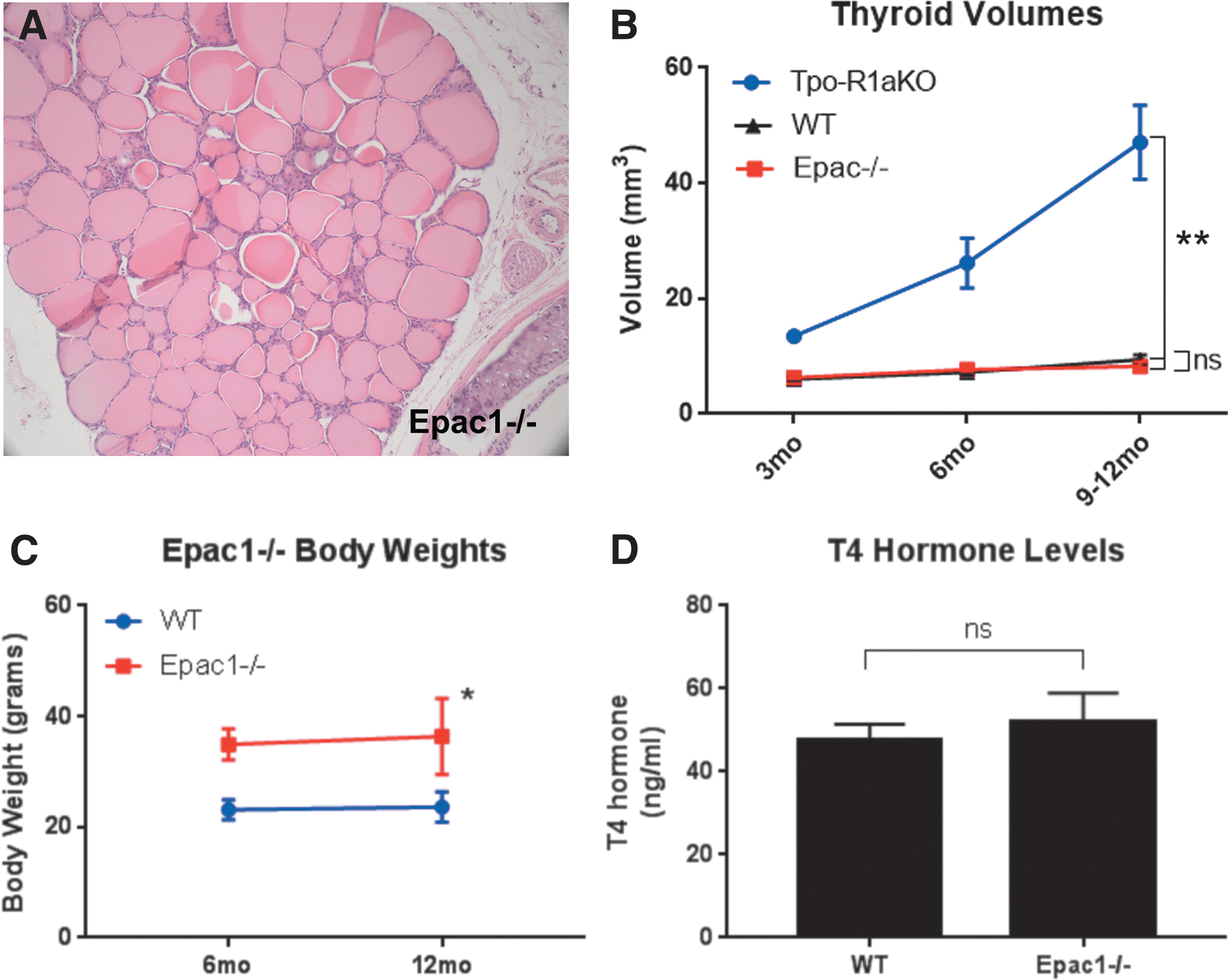
Epac1 is expressed in follicular cells of WT and Tpo-R1aKO thyroid tumors, as shown by immunohistochemistry and Western blot (Fig. 4A–C). Global Epac1–/– mice (25) were obtained to aid in the elucidation of signaling mediators in the PKA-Rap1 carcinogenic cascade. Epac2 was not found to be expressed at detectable levels in the tumors (Fig. 4C and D), nor did Epac2 expression increase upon deletion of Epac1 (Fig. 4D). Epac1–/– thyroids exhibit normal histological features (Fig. 5A) and growth patterns, with volumes similar to WT littermate thyroids (8.26 mm3 for Epac1–/– vs. 9.41 mm3 for WT; p = 0.3915; Fig. 5B). Although Epac1–/– females (n = 4) were on average larger than their WT littermates (n = 4; 36.38 vs. 23.6 g at one year of age; p = 0.013; Fig. 5C), T4 levels were similar to WT animals (Fig. 5D). Therefore, the increased body weight did not seem to be related to altered thyroid function.
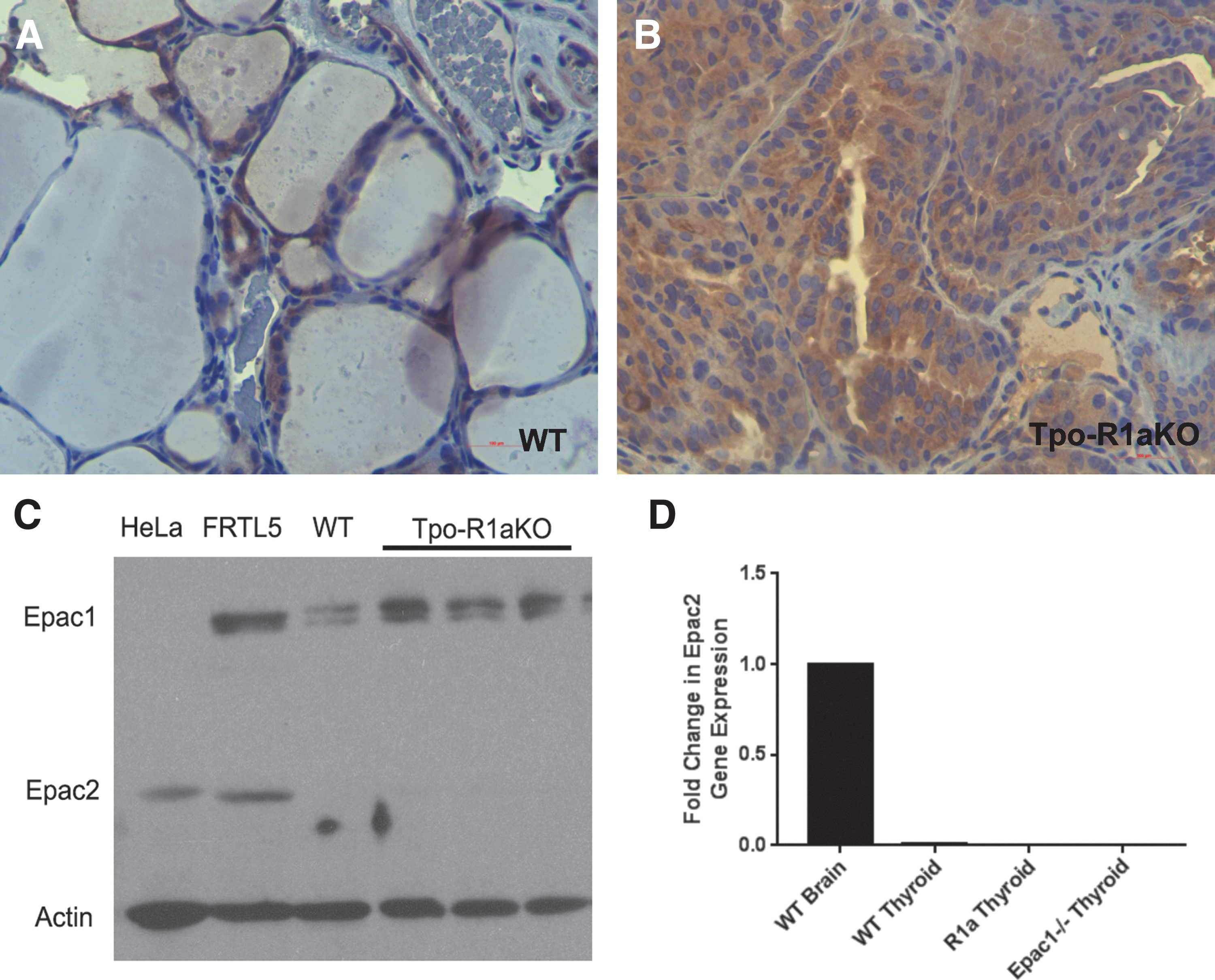
Epac1 is expressed in Tpo-R1aKO tumors. (
Epac1
–/– mice have normal thyroids. (
To determine whether Epac1 is involved in activation of Rap1 in Tpo-R1aKO mice, Epac1–/– mice were crossed with Tpo-R1aKO animals to produce Tpo-Cre;Prkar1aloxP/loxP;Epac1–/– mice, hereafter referred to as TpoR1a-EpacKO. Ultrasonography revealed that thyroids from TpoR1a-EpacKO mice had volumes comparable to littermate Tpo-R1aKO mice at all time points studied (Fig. 6A–D; 40.87 mm3 for TpoR1a-EpacKO vs. 38.02 mm3 for Tpo-R1aKO at nine months of age; p = 0.6397). Histological analysis also revealed similar incidences of carcinomas between the two groups (81.82% for Tpo-R1aKO vs. 85.71% for TpoR1a-EpacKO at nine months of age; p = 0.7737). These data suggest that Rap1 is functioning downstream of PKA independent of Epac1.
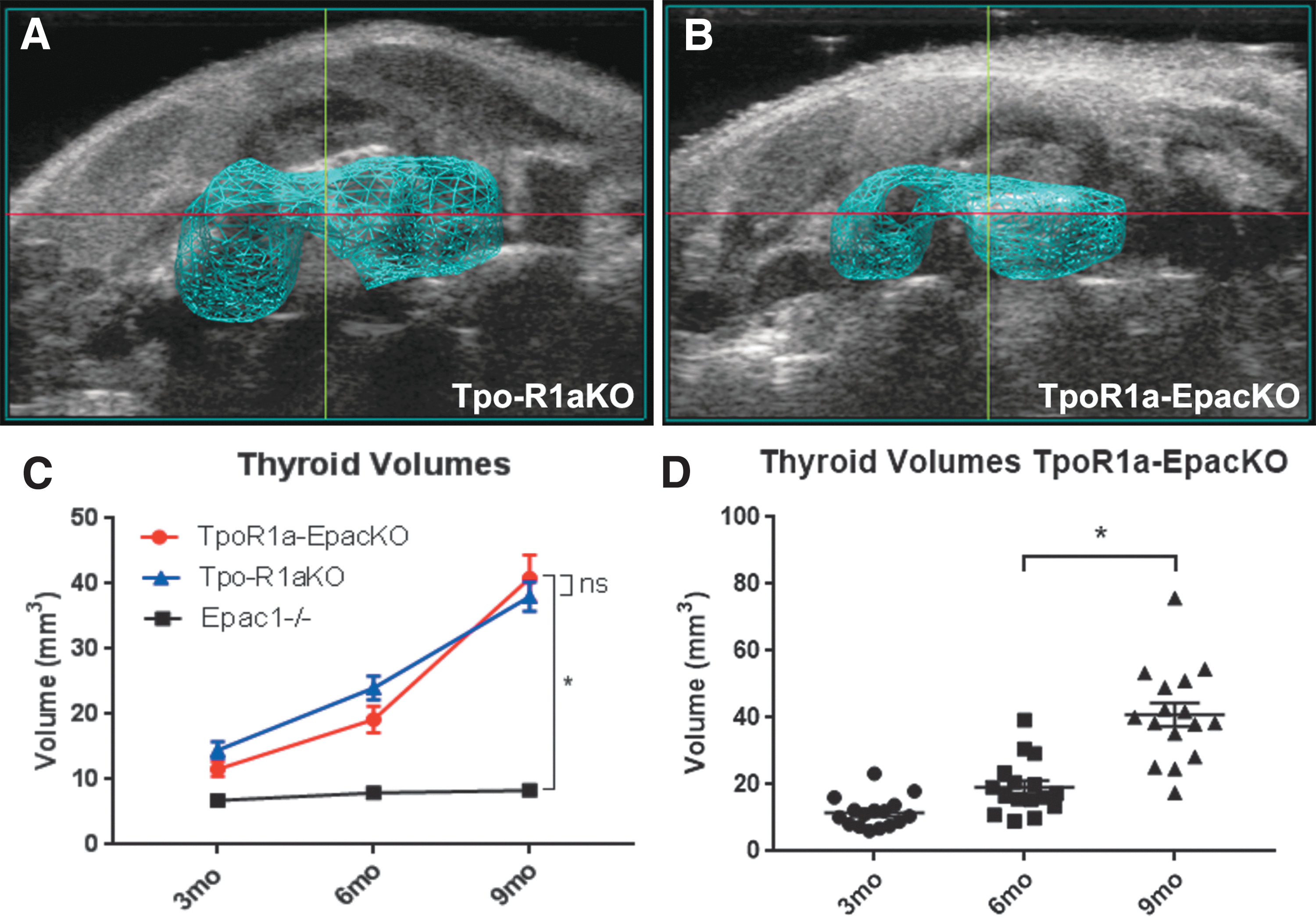
Epac1 is not required for tumor formation in TpoR1aKO mice. (
To probe the relative roles of Epac1 and PKA in this model further, Epac1–/– and WT littermates were treated with a goitrogenic protocol (9) to cause hypersecretion of TSH (Fig. 7A) and activation of the cAMP pathway upstream of PKA. After six months of treatment, the animals were sacrificed, and thyroids were collected for histological analysis. It was found that both groups developed thyroid hyperplasia that was histologically indistinguishable from one another (Fig. 7B and C), indicating that Epac1 likewise does not play a role in chronic TSH-mediated thyroid cell proliferation.
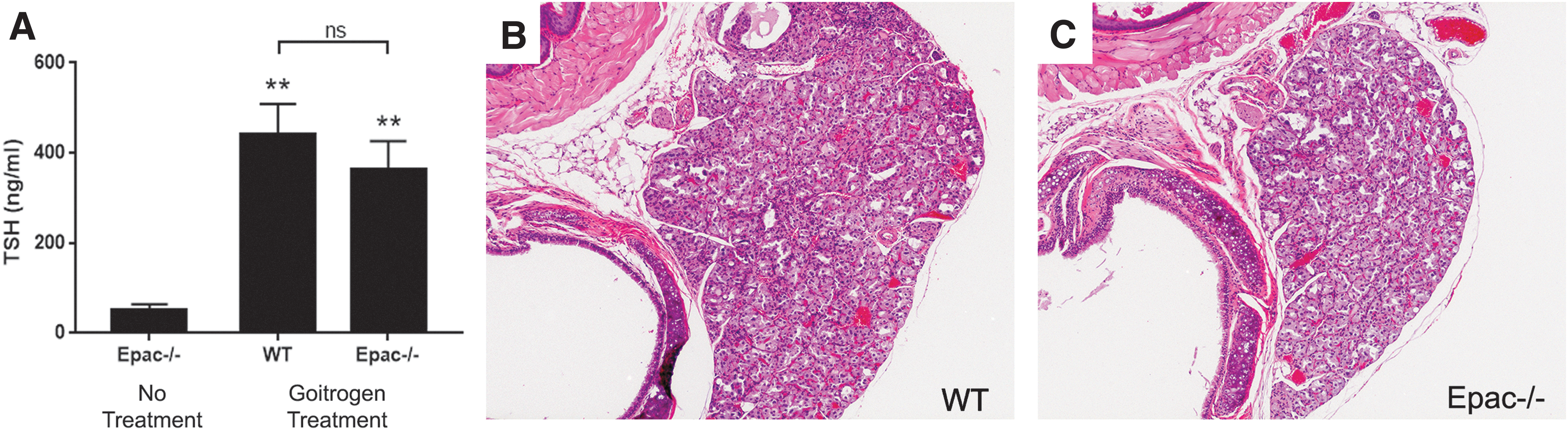
Epac1 is not involved in thyrotropin (TSH)-mediated thyroid hyperplasia. (
Discussion
As incidence rates of thyroid cancer have continued to rise at the fastest rate of all malignancies, the study of signaling pathways involved in thyroid tumorigenesis remain relevant to aid in the development of novel therapeutics. This study observed that Rap1 plays an important role in thyroid carcinogenesis arising from over-activation of the PKA pathway. Using targeted-deletion mouse modeling, it was determined that ablation of Rap1, specifically the Rap1b isoform, reduces the risk of developing thyroid cancer as a result of Prkar1a KO.
Previous work has established an oncogenic role of Rap1b resultant from cAMP stimulation. Although Rap1b activation alone is not sufficient to produce thyroid cancer in mice, it is still thought to be a major player in mitogenic signal transduction, as its activity is associated with many human cancers, including thyroid cancer (9,14 –17). Using an established mouse model of PKA-dependent follicular thyroid cancer, the function of Rap1 in in vivo tumor development was probed. It was found that deletion of Rap1a and Rap1b in the setting of PKA over-activation resulted in a 65% reduction in incidence of thyroid carcinoma. Further, isoform specific knockout revealed that Rap1b deletion alone produced similar results to deleting both isoforms together, indicating that Rap1b is important for tumor formation. Although Rap1 ablation was sufficient to suppress tumor formation significantly, the carcinogenic phenotype was not completely rescued, implying that more complex signaling interactions may be at play.
Previously, interactions between PKA, Rap1, and Epac1 during thyroid carcinogenesis were unclear. Some studies have shown that both PKA and Epac signal to Rap1 downstream of TSH (13,18). However, the nature of these interactions seems to be cell context-dependent (21,22). Here, it was demonstrated that PKA is acting on downstream target Rap1 to promote in vivo thyroid carcinogenesis independently of Epac1, as deletion of Epac1 in a Prkar1a KO background had no effect on tumor growth. To discern further whether Epac1 may have an antagonistic relationship with PKA, the cAMP pathway was stimulated upstream of PKA in WT and Epac–/– mice by inducing secretion of excess TSH through treatment with antithyroid drugs. As no significant differences were observed between groups, it was concluded that Epac1 is not directly involved in PKA-Rap1-associated thyroid tumorigenesis. These findings are supported by others who have shown that PKA can directly regulate Rap proteins using a specific phosphorylation site at serine 180 on Rap1a and serine 179 on Rap1b (27). Phosphorylation of Rap by PKA has been shown to regulate its subcellular localization, as well as its downstream action, including activation of certain downstream effectors such as ERK and Rap-dependent regulation of cell migration (28,29). These previously published data show that PKA has a direct mechanism of controlling Rap action and downstream cellular processes and, taken together with the present results, suggest that the PKA-Rap1 pathway can operate independently of Epac1 in thyroid cancer initiation and progression.
The functional role of Epac1 in tumorigenesis remains elusive. Despite robust upregulation of Epac1 expression in Tpo-R1aKO mice, these studies do not reveal a causative role for Epac in PKA-mediated thyroid tumor formation. Numerous in vitro studies have also noted increased Epac1 expression under tumor promoting conditions. However, similarly to the present observations in vivo, Rap1 activation was not dependent on Epac1 in most thyroid cancer cell lines (20,30). These findings do not exclude an indirect or supportive role of Epac1 in tumor growth, rather the often-opposing results of PKA/Epac-Rap pathway activation in different cell types is consistent with this theory.
In conclusion, the data indicate that PKA signaling to Rap1b is a key signaling node for follicular thyroid carcinogenesis. Epac has been shown to activate Rap proteins in multiple systems, although the data presented here indicate that Epac activity is dispensable for thyroid tumorigenesis in vivo. Interestingly, although they are both signaling molecules implicated in the TSH/cAMP/PKA pathway, neither Epac nor Rap1 appear to be required for normal thyroid morphology or function. These studies should help shed further light on oncogenic signaling.
Footnotes
Acknowledgments
We would like to thank Alexei Morozov for sharing the Rap1aloxP/loxP;Rap1bloxP/loxP mouse model, and Xiaoli Zhang for statistical analyses. This work was supported by NIH R01CA170249, P01CA124570 (L.S.K.)m and a Pelotonia Postdoctoral Fellowship Award (D.H.). A.A. and A.M. were also supported Pelotonia fellowships for graduate students and undergraduates, respectively. The Hormone Assay and Analytical Services Core at Vanderbilt University is supported by NIH DK059637 and DK020593.
Author Disclosure Statement
No competing financial interests exist.