
Abstract
Background:
Thyroid hormone receptors (TRs) are tightly regulated by the corepressors nuclear receptor corepressor (NCoR) and silencing mediator of retinoic acid and thyroid hormone receptors. Three conserved corepressor/NR signature box motifs (CoRNR1–3) forming the nuclear receptor interaction domain have been identified in these corepressors. Whereas TRs regulate multiple normal physiological and developmental pathways, mutations in TRs can result in endocrine diseases and be associated with cancers due to impairment of corepressor release. Three mutants that are located in helix H11 of TRs are of special interest: TRα-M388I, a mutant associated with the development of renal clear cell carcinomas (RCCCs), and TRβ-Δ430 and TRβ-Δ432, two deletion mutants causing resistance to thyroid hormone syndrome.
Methods:
Several cell-based and biophysical methods were used to measure the affinity between wild-type and mutant TRα and TRβ and all the CoRNR motifs from corepressors to quantify the effects of different thyroid hormone analogues on these interactions. This study was coupled with the measurement of interactions between wild-type and mutant TRs in the context of a heterodimer with RXR to a NCoR fragment in the presence of the same ligands. Structural insights into the binding mode of corepressors to TRs were assessed in parallel by nuclear magnetic resonance spectroscopy.
Results:
The study shows that TRs interact more avidly with the silencing mediator of retinoic acid and thyroid hormone receptors than with NCoR peptides, and that TRα binds most avidly to S-CoRNR3, whereas TRβ binds preferentially to S-CoRNR2. In the studied TR mutants, a transfer of the CoRNR-specificity toward CoRNR1 was observed, coupled with a significant increase in the binding strength. In contrast to 3,5,3′-triiodothyronine (T3), the agonist TRIAC and the antagonist NH-3 were very efficient at dissociating the abnormally strong interactions between mutant TRβs and corepressors. A strong impairment of T3-binding for TRβ mutants was shown compared to TRIAC and NH-3 and could explain the different efficiencies of the different ligands in releasing corepressors from the studied TRβ mutants. Consequently, TRIAC was found to be more effective than T3 in facilitating coactivator recruitment and decreasing the dominant activity of TRβ-Δ430.
Conclusion:
This study helps to clarify the specific interaction surfaces involved in the pathologic phenotype of TR mutants and demonstrates that TRIAC is a potential therapeutic agent for patients suffering from resistance to thyroid hormone syndromes.
Introduction
H
Like other NRs, TRs are composed of a DNA-binding domain (DBD), linked by a hinge to the ligand-binding domain (LBD), which carries the transcriptional regulation function (AF-2). In the absence of T3, TRs are bound to the DNA response elements of their target genes and recruit corepressor complexes. Corepressors further recruit various proteins, including histone deacetylases (HDACs), forming a repressor complex that condenses chromatin and therefore prevents gene expression on positively regulated genes. Binding of thyroid hormone (T3) to the LBD of TR leads to the destabilization of the corepressor interface and the recruitment of coactivators such as the steroid receptor coactivator-1 (SRC1). Coactivators will further recruit a larger activator complex containing histone acetyl transferases (HATs) that will loosen the chromatin and allow the transcription of positively regulated target genes (3).
Two main corepressors—the nuclear receptor corepressor (NCoR) (4) and the silencing mediator of retinoic acid and thyroid hormone receptors (SMRT) (5)—have been described as NR partners (Fig. 1A). NCoR and SMRT share a common organization and interact with NRs through their receptor interaction domains (RIDs), located in the carboxy-terminal part. RIDs are around 300 amino acids long and contain three so-called corepressor/nuclear receptor boxes (CoRNR1–3) that mediate the direct interaction with NRs and all contain a (L/I)xx(V/I)/I consensus motif (3,6
–10). Whereas the mode of interaction of corepressors with TR has not yet been precisely determined, their interaction with others NRs has been described for two of these boxes—CoRNR1 and CoRNR2— revealing very specific and different modes of binding (Fig. 1B and Supplementary Table S1; Supplementary Data are available online at
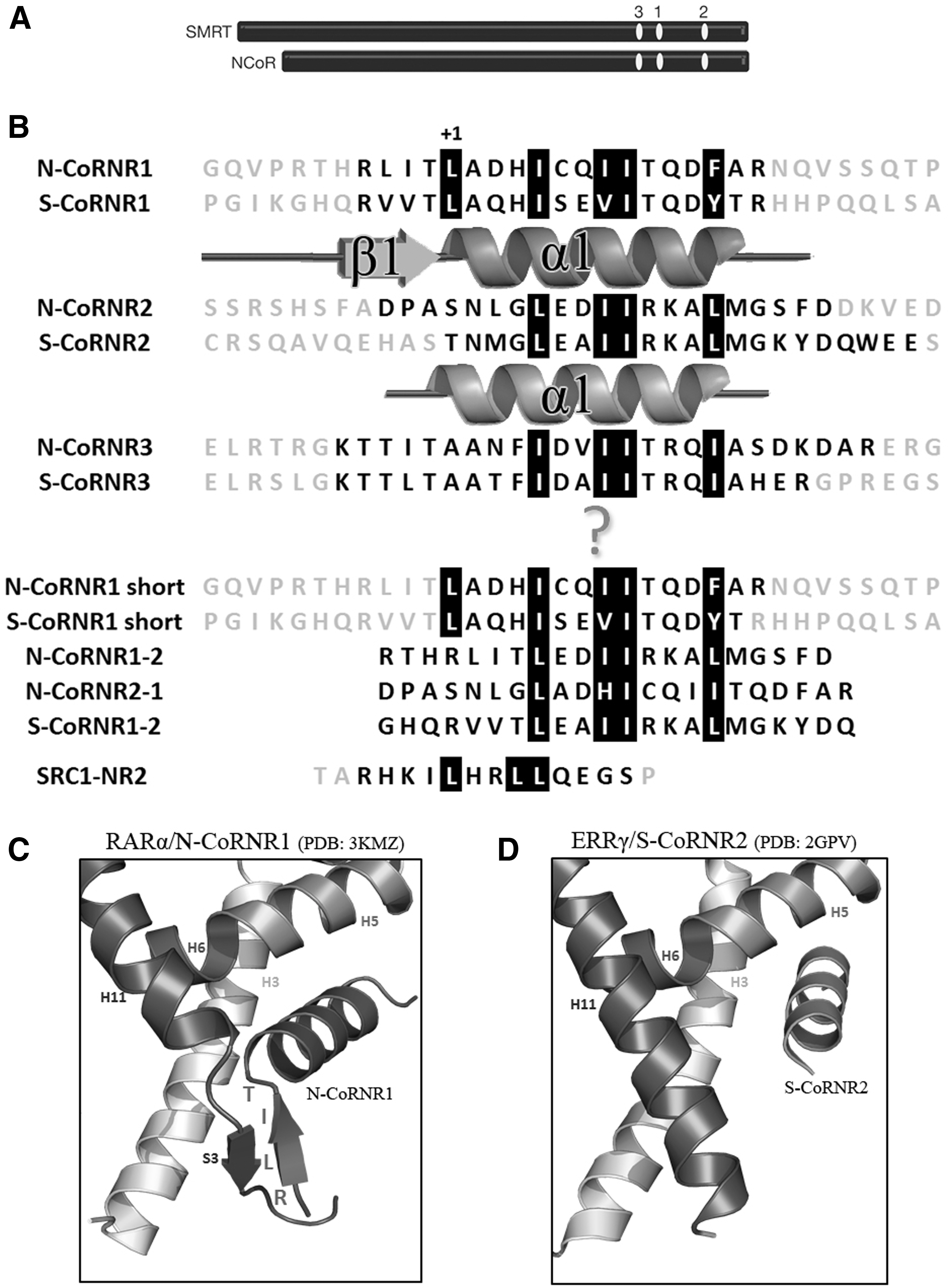
CoRNR boxes of corepressors. (
Due to the different binding modes of CoRNR1 and CoRNR2 with NRs, the nature of the interactions is different: CoRNR1 interacts specifically with high affinity with a subtype of NRs that have strong constitutive repressive function, whereas CoRNR2 interacts more broadly and with lower affinity with other NRs. In contrast, the CoRNR3 binding mode is still unknown, as no structure of a complex between NR and CoRNR3 has been solved until now.
Several endocrine diseases and cancers have been linked to aberrant interactions between mutant forms of TRs and corepressors. Unfortunately, many of the currently available drugs shown to treat these diseases more or less successfully have proven to have serious side effects that have dramatically limited their clinical usefulness (18). Mutations in THRβ are the most common cause of resistance to thyroid hormone (RTH) syndrome, an inherited human endocrine disorder manifested as a diminished responsiveness to T3 (19,20). In contrast, RTH alpha due to THRα gene mutations is less common but probably under-recognized (21). RTH TR mutants operate as dominant-negative inhibitors of wild-type TR function, as these mutations impair corepressor release. In addition, a particular high rate of TR mutations has been found in human renal clear cell carcinomas (RCCCs)—cancers that account for 90% of renal cancers. The majority of the RCCC TR mutants are defective for transcriptional activation and behave as dominant-negative inhibitors of wild-type TR function (22). Although only a subset of the dominant-negative RCCC and RTH TR mutants display impaired hormone binding, all fail to release corepressors appropriately in response to T3, a trait that closely correlates with their defective transcriptional properties. Interestingly, among the identified mutations, two deletions (TRβ-Δ430 and TRβ-Δ432) and a point mutation (TRα-M388I) occur in the region of helix H11 of TR. As it has already been shown that a secondary structure transition from β-strand S3 to α-helix H11 is the master regulator of corepressor dissociation in NRs acting as transcriptional repressors (15), in this study, it was hypothesized that the mutations TRβ-Δ430, TRβ-Δ432, and TRα-M388I could induce a transition from the α-helix (H11) to a β-strand (like S3 from RAR) to allow strong interaction of the mutated TRs with corepressors.
The aim of the present study was to provide molecular insights into the mechanism of wild-type TR–corepressor association and the impaired T3-induced corepressor release from TR mutants. Using a combination of biochemical, biophysical, cell-based, and structural approaches, it was found that CoRNR2 and CoRNR3 interactions with wild-type TRs seem to involve an helix interaction similar to that observed for CoRNR2 with others NRs. In contrast, all three mutations shift the CoRNR box specificity toward CoRNR1 by inducing an increase of the binding affinity of these TR mutants for this corepressor motif. Several lines of evidence suggest that the mechanism involves a mutation-induced stabilization of a β-sheet interaction between CoRNR1 and TR, similar to that seen in the RARα– and RevErb–CoRNR1 complexes. Very interestingly, it was found that contrary to T3, the TR agonist TRIAC and the antagonist NH-3 are able to dissociate the pathologic interactions between mutant TRβ and corepressors. In addition, TRIAC was found more effective than T3 in decreasing the dominant activity of TRβ-Δ430 by facilitating coactivator recruitment. This finding may help in the development of therapies using thyroid hormone analogues for the treatment of RTH syndromes and RCCCs.
Methods
Ligands and peptides
3,3′,5-triiodo-L-thyronine was purchased from Sigma–Aldrich. The fluorophore-labeled coactivator and corepressor peptides were purchased from EZbiolab. Figure 1 summarizes the sequences of the peptides used.
Protein purification
Human TRα-LBD (E159-D407) and TRβ-LBD (E213-D461) were expressed in BL21 DE3 Escherichia coli strain (Invitrogen) using the pET15b vector. Protein expression was induced from five hours to overnight at 18–23°C with 1 mM isopropyl-β-D-thiogalactopyranoside. Bacteria were pelleted and lysed by sonication in lysis buffer (20 mM Tris, pH 7.5, 500 mM NaCl, 10% [v/v] glycerol, 3 mM NOG, 25 mM imidazole). Proteins were purified with a His-Trap HP (GE Healthcare) followed by a Superdex 75 column (GE Healthcare) and put in final buffer (20 mM Tris, pH 7.5, 200 mM NaCl, 5% [v/v] glycerol, 1 mM EDTA, 5 mM DTT). Mutant TRα-M388I, TRβ-Δ430, and TRβ-Δ432 gene constructions were generated as in Rosen and Privalsky (22) and purified as described above. The heterodimers RXRα-TRα LBD, RXRα-TRαM388I LBD, RXRα-TRβ LBD, RXRα-TRβ-Δ430 LBD, and RXRα-TRβ-Δ432 LBD were prepared by incubation of each TR LBD with an equimolar amount of purified hRXRα LBD (amino acids 225–462) prepared as in Fattori et al. (23) followed by a size exclusion chromatography on a S75 superdex gel filtration column equilibrated in a buffer consisting of 50 mM Tris, pH 7.5, 150 mM NaCl, 5% (v/v) glycerol, and 2 mM DTT. Fractions corresponding to the heterodimers were pooled, and the stoichiometry of the heterodimers was checked with sodium dodecyl sulfate polyacrylamide gel electrophoresis. The NCoRNID protein corresponds to the sequence from Q2059 to E2325 of mouse N-CoR. This sequence was inserted into a modified pET15 vector (kindly given by Marc Ruff, IGBMC, Strasbourg, France) allowing the expression of the NCoRNID fragment fused to a 6His-thioredoxin tag. The protein was purified by using a nickel affinity column in 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 2 mM DTT, followed by a Tev cleavage of the tag. A final purification step was then performed with size exclusion chromatography on a S75 superdex gel filtration column in the same buffer.
Thermal shift assays (Thermofluor®)
This method measures protein unfolding based on fluorescence detection of the denatured form of the protein (24). Solutions of 25 μL containing 5 μM protein, in the presence of dimethyl sulfoxide (condition without ligand) or in the presence of various amounts of ligands in excess, and 5 × Sypro® Orange in 50 mM Tris, pH 8.0, 200 mM NaCl were added to the wells of a 96-well polymerase chain reaction plate. The plates were sealed with an optical sealing tape (Bio-Rad) and heated in a 7500 Real Time PCR system (Applied Biosystems) from 25°C to 95°C at 1°C intervals. Fluorescence changes in the wells were monitored with a photomultiplier tube. The wavelengths for excitation and emission were 545 and 568 nm, respectively. The melting temperatures (T m) were obtained by fitting the fluorescence data with a Boltzmann model using GraphPad Prism.
Fluorescence anisotropy measurements
Measurements of the binding affinities of the fluorescent peptides for TR LBD in the presence or absence of ligands was performed using a Safire2 microplate reader (TECAN). For the fluoresceine-labeled CoRNRs, the excitation wavelength was set at 470 nm, and emission was measured at 530 nm. For the rhodamine-labeled CoRNRs, the excitation wavelength was set at 530 nm, and emission was measured at 580 nm. The reported data are the average of at least three independent experiments, and error bars correspond to standard deviations. The buffer solution for assays was 20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 5 mM DTT, and 10% (v/v) glycerol. The measurements were initiated at 40 μM or 20 μM of protein, and the sample was then diluted successively by a factor of 2 with the buffer until the lowest protein concentration at 9.7 nM. Fluorescent peptides were added to protein samples at 4 nM, allowing establishment of the titration curve. Ligands, when added, were at a final concentration of three molar excess relative to the highest concentration of protein.
The ligand titration assay of the complexes between N-CoRNR1 (labeled with fluoresceine) and TRβ-Δ430 or TRβ-Δ432 with the three ligands T3, TRIAC, and NH-3 was performed using a CLARIOstar microplate reader (BMG Labtech) with excitation and emission wavelengths of 480 and 520 nm, respectively. Ligands, initially at a concentration of 160 μM, were serially diluted 1:2 fifteen consecutive times into the same buffer solution as above in a volume of 25 μL, directly into a 384-well black-walled microplate NBS (Corning). Then, 25 μL of a mix of protein at 2 μM and N-CoRNR1 at 8 nM were distributed in each condition. After 30 minutes of incubation in the dark, fluorescence polarization was read.
All fluorescence anisotropy measurements were done at least in triplicate. Experimental values are shown as the mean ± standard deviation and were analyzed by nonlinear regression with an equation for sigmoidal dose response (variable slope) in GraphPad Prism.
Microscale thermophoresis
A fluorescent probe (Atto647N maleimide; Invitrogen) was attached to the thiol group of the sole cysteine of the N-CoRNID fragment, according to the manufacturer's instructions. This cysteine (C2074) is in the CoRNR1 motif but is not involved in the interaction between RAR and N-CoR, as it points to the outside in the crystal structure of the complex (15). RXRα-TRs and TRs LBDs were prepared as a twofold serial dilution in microscale thermophoresis (MST) buffer (NanoTemper Technologies GmbH) and added to an equal volume of 40 nM labeled N-CoRNID. After 10 minutes of incubation time, the complex was filled into Monolith NT.115 Premium Coated Capillaries (NanoTemper Technologies GmbH), and thermophoresis was measured using a Monolith NT.115 Microscale Thermophoresis device (NanoTemper Technologies GmbH) at an ambient temperature of 22°C, with 5 s/30 s/5 s laser off/on/off times, respectively. Instrument parameters were adjusted with 20% red LED power and 20% IR-laser power. Data from three independent measurements were analyzed (NT Analysis software last version; NanoTemper Technologies GmbH) using the signal from Thermophoresis + T-Jump.
Nuclear magnetic resonance spectroscopy
2D Total Correlated SpectroscopY (TOCSY; mixing time 60 ms) (25,26), NOESY (mixing time 200 and 500 ms) (26,27), 13C–1H Heteronuclear Single Quantum Coherence (HSQC), and TOCSY–HSQC (mixing time 60 ms) were recorded on a 700 MHz Bruker Avance spectrometer equipped with a cryogenic H/C/D/N probe with a Z-axis gradient at 283 and 293 K to obtain the resonance assignment of all free peptides, at a concentration ranging from 200 to 400 μM. Proton and carbon chemical shifts were referenced relative to 2,2-dimethyl-2-silapentane-5-sulfonic acid resonances.
All saturation transfer difference (STD) experiments (28), except for S-CoRNR1, were recorded on a 500 MHz Bruker Avance spectrometer equipped with a cryogenic H/C/D/N probe with a Z-axis gradient at three temperatures (274, 278, and 283 K). The STD experiments for S-CoRNR1 were recorded on a Bruker 950 MHz equipped with a cryogenic H/C/D/N probe with a Z-axis gradient at 275, 278, and 283 K. All experiments were done with wild-type TRα, except for N-CoRNR3 for which wild-type TRβ was used. The choice of this isoform was dictated by the need to maximize the STD intensities, which are proportional to the dissociation rate of the complex (k off). All spectra were recorded with a protein:peptide ratio of 1:20, with peptide concentrations ranging from 400 μM (N-CoRNR2) to 200 μM (S-CoRNR3) and 100 μM (S-CoRNR1). A saturation transfer of 2 s was achieved with equally spaced 50 ms Gaussian shaped pulses (separated by a 1 ms delay) centered at 0.1 ppm. Subtraction of three induction decay values with on- and off-resonance protein saturation was achieved by phase cycling. Experiments were recorded with 128 dummy scans between 8192 and 24,572 transient and a relaxation delay of 2.5 s (Aq + D 1). All nuclear magnetic resonance (NMR) spectra were processed and analyzed using Gifa (29), Nmrview 5.0 (30), and Topspin 2.6 (Bruker Biopsin). All structure calculations were performed with Aria 2.2 (31) interfaced with the CNS program (32).
The NOE cross peaks were volume integrated by using the routine in NMRView. In addition, dihedral angle restraints were obtained by using the TALOS-N program (33) with HN, Hα, 13Cα, and 13Cβ chemical shifts as input, after verification that inclusion of chemical shift–derived constraints did not alter the overall fold but only accelerated the convergence, quality, and precision of structure calculations. The constraints were introduced for structure calculation with the standard protocol of Aria 2.2, with 100 structures calculated per iteration, of which the 20 best structures were selected for further evaluation.
Modeling of TR–corepressor complexes was done using the Modeller software using different crystallographic structures of NRs complexed with corepressor RIDs as templates (34,35). Protons were added with the Whatif server (36). As in all crystallographic structures used as templates (PDB codes 3H52, 2OVM, 2OVH, 4WVD, 1KKQ for CoRNR2, and PDB codes 3KMZ and 3N00 for CoRNR1 complexes), the electronic density of amino- and carboxy-terminal parts of the corepressors was only partially visible, five models of TR–CoRNR complexes were generated in order to get a view of the plausible conformational space for these missing parts. The STD effects were simulated for each model.
Back calculation of STD intensities was calculated with CORCEMA-ST v3.8 (37). An order parameter value of 0.85 for methyl groups, and a K on value of a 108/s value were used, with the association constants set to the experimental value measured by fluorescence anisotropy. The correlation times were set to 0.5 and 13 ns for the free and bound states, respectively. Calculations with different correlation time values exploring the 0.2–2 and 10–30 ns for the free and bound forms, respectively, showed that the simulated profiles were much more dependent on the template model chosen than on the correlation time values used.
Results
Corepressor binding modes to TRα and TRβ
First, the CoRNR binding specificity was assessed by measuring the dissociation constants (Kds) between wild-type TRs (TRα and TRβ) and the six fluorescein-labeled CoRNRs from N-CoR (N-CoRNR1–3) and SMRT (S-CoRNR1–3; Figs. 1 and 2A and B, Supplementary Fig. S1, and Table 1). Both TRα and TRβ interact not only with NCoR peptides but also with SMRT peptides, in some cases even more efficiently; the highest affinities toward TRα and TRβ were observed with S-CoRNR3 and S-CoRNR2 peptides, respectively (Fig. 2A and B).

Affinities of wild-type ([
Dissociation Constants (Kd in μM) Measured from Fluorescence Anisotropy Titrations Between Fluorophore-Labeled Corepressor Peptides and hTR-LBD Constructs in the Absence or Presence of Triiodothyronine
CoRNR, corepressor/nuclear receptor; T3, triiodothyronine.
In order to get structural insights into the interaction modes of the different CoRNRs with TRs, and as all attempts to crystallize TRs in the presence of CoRNR boxes have failed, the NMR STD technique was used. In favorable cases, STD enables not only the identification of the interaction surface of the small molecular weight partner in a complex, but also provides information about its most probable structure (37). For all CoRNR types, STD spectra were recorded in the presence of the unliganded form of wild-type TR to identify the interaction surface of the CoRNR boxes. Figure 3 depicts the experimental data for the three different types of CoRNRs.

Experimental saturation transfer difference (STD) profiles for CoRNR–TR complexes. STD recorded at three different temperatures are plotted as a function of the residue number of each CoRNR motif, following the numbering adopted in Figure 1. (
S-CoRNR1, N-CoRNR2, and S-CoRNR3 present different STD experimental profiles: whereas the maximal intensity is observed for residues at position 0, +5, +10, and +14 for S-CoRNR1, maximal STD effects are observed at position +3, +5, +8, +9, and +14 for N-CoRN2 and +3, +5, +8, +9, +11, and +13 for S-CoRNR3. Hence, the amino-terminal part of CoRNR1, but not that of N-CoRNR2 and S-CoRNR3, seems to be part of the interaction surface with TR. It is important to note that a similar profile was obtained for N-CoRNR1 as for S-CoRNR1 but only at 275 K due to the lower stability of the former peptide (Supplementary Fig. S2). To explain the interaction between TRs and the amino-terminal CoRNR1 motifs, it was hypothesized that TRs could adopt a β-strand conformation in place of helix H11, similar to S3 in RARα (15), enabling the formation of a β-sheet interface between the amino-terminal β1 strand of CoRNR1 and the β-strand S3 of TRs. To validate this hypothesis, the theoretical STD profiles were simulated according to the different binding modes.
As can be seen in Figure 4, the experimental STD effects measured on the TRα–CoRNR1 complex are more in accordance with the STD effects back calculated from a template structure containing a N-CoRNR1 peptide (PDB code 3KMZ) (15), whereas those measured on the TRα–CoRNR2 complex fit well with the ones simulated on a complex containing a CoRNR2 peptide (PDB code 4WVD) (38). This was true irrespective of the template chosen for the modeling of TR–CoRNR1 (PDB codes 3KMZ) and 3N00) (15,16) or of TR–CoRNR2 (PDB codes 2OVM and 2OVH, 4WVD, and 3H52) (14,38,39), although some structures were in better accordance than others. Thus, the comparison between experimental and simulated STD data confirms that the mode of interaction in β-sheet observed between RevErb and RARα receptors (15,16) and the CoRNR1 box of corepressor can be extended to TR as well.
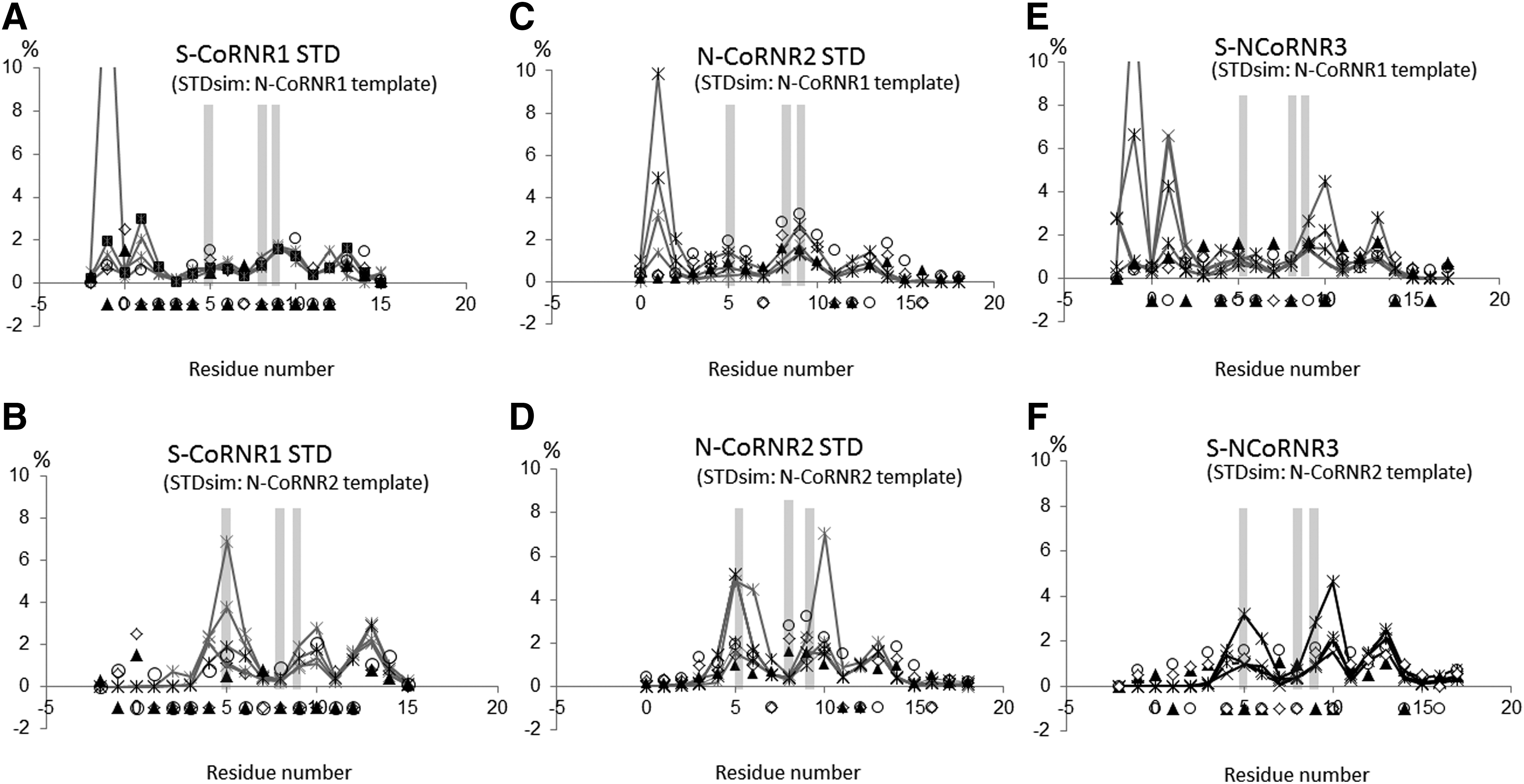
Comparison of experimental and simulated STD effects. Experimental STDs were measured on complexes between (
The experimental STD profile measured on the TRβ–CoRNR3 complex is more in line with STD intensities simulated from structures containing a CoRNR2 peptide than a CoRNR1 peptide (Fig. 4), although the amino-terminal part of S-CoRNR3 seems to interact more intimately with TR than the one of N-CoRNR2. There is a modest but non-negligible increase of STD effects for residues belonging to the S-CoRNR3 amino-terminal part that could reflect a different positioning of helix H12 with respect to the peptide, as seen for example in a crystal structure with the S-CoRNR2 (PDB code 1KKQ) (11). In the former template structure, the position of helix H12 forces the amino-terminal part of the corepressor peptide in the different complex models to fold back near helix H11, resulting in slightly higher STD effects than in models generated from the 4WVD template (Supplementary Fig. S3). Alternately, it could also originate from a difference in the proton relaxation times of the free N-CoRNR2 and S-CoRNR3 peptides, since different dynamics can impact on STDs (37). To characterize the interaction mode of CoRNR3 further, structural information on this motif was gained by doing NMR on its free form. Analysis of the 1H and 13C chemical shifts with TALOS (33) and inspection of NOESY map indicated that S-CoRNR3 adopts random and dynamical conformations in solution (Supplementary Fig. S4). However, the presence of a seemingly continuous stretch of weak NHi/NHi+1 NOEs covering the sequence from −1 to +17 was noted. No other NOE indicative of helix fold or turn (such as NHi/NHi+2, αHi/NHi+2, αHi/NHi+3, αHi/Hβi+3) could be observed for this peptide. This observation suggests the presence of a transient helix present throughout the CoRNR3 peptide sequence. Therefore, all the data argue against the hypothesis of a β-strand formation in the amino-terminal part of CoRNR3 peptides, suggesting a binding mechanism more similar to that of CoRNR2 than of CoRNR1. For further validation, similar NMR data were recorded on free CoRNR1 and CoRNR2 peptides. Evidence was also found (stretches of NHi/NHi+1 NOEs) of the presence of a transient helix for the CoRNR1 peptides, but limited to their central and carboxy-terminal parts (starting from residue 2 and 3 for N- and S-CoRNR1 peptides, respectively). Interestingly, the position of these transient helixes corresponds to the location of the structured helixes observed in their complexes with RARα (15) and RevErb (16). Similarly, a NOE set characteristic of a well-formed helix was found for the free N-CoRNR2 peptide. The completeness of the NMR restraint set was sufficient to calculate a structure of this peptide that proved to be very similar to the one found in its complex with FXR (38) (Supplementary Fig. S5).
Together, these results suggest that wild-type TRα and TRβ interact with each of the three CoRNR motifs of corepressors using different binding modes. The NMR experiments provide strong evidence that TRs could adopt a β-strand carboxy-terminal of helix H10 to interact with CoRNR1 and that the interaction of TRs with CoRNR2 and CoRNR3 motifs are similar to one another, involving mainly interaction with the helix part of these two motifs.
TRα and TRβ mutations alter corepressor binding
Next, the impact of pathologic mutations on the interactions of mutant TRs with corepressors was analyzed. A general trend associated with these mutations is an increased affinity of most peptides for all TR variants, however with some subtype specificities (Fig. 2C–E, Supplementary Fig. S1, and Table 1). When compared to TRα, the binding affinity of TRα-M388I for all NCoR peptides and S-CoRNR1 is enhanced by a factor of 2, whereas it decreases slightly with S-CoRNR2 and S-CoRNR3 (Fig. 2C and Table 1). In contrast, both TRβ mutations (Δ430 and Δ432) show a greater effect on CoRNR interactions. The deletions Δ430 and Δ432 increase the binding strength of TRβ mutants to N- and S-CoRNR1 by a factor of 10 and 4, respectively, whereas TRβ-Δ432 has a five times higher affinity for S-CoRNR3 than TRβ (Fig. 2D and E and Table 1). Only N-CoRNR2 shows a slightly decreased affinity for TRβ-Δ432. However, overall, the most remarkable effect that could be observed with these TR mutations is the clear shift of the binding specificity toward CoRNR1, which becomes the preferred CoRNR for all TR variants. Interestingly, this shift in the binding specificity relies mostly on the strong increase of the affinity of CoRNR1 motif for all TR variants rather than on a decrease of the interaction strength of other CoRNRs.
Using MST, next, the affinity of a NCoR RID fragment (named NCoRNID hereafter) was measured for all TR strains in the context of heterodimers with RXRα (Table 2 and Supplementary Fig. S6). Unlike SMRT, it was possible to design a stable NCoRNID, containing CoRNR1 and CoRNR2, which could be purified to homogeneity. Attempts to produce larger corepressor fragment containing the three CoRNR boxes were unsuccessful. The data reported in Table 2 and Supplementary Figure S6 confirm that TRβ-RXRα heterodimers display a slightly higher affinity for NCoR than TRα-RXRα heterodimers. Moreover, they show that TR-RXRα heterodimers interact generally better with the RID than with individual peptides, suggesting an involvement of additional contacts, likely mediated by RXRα, in their interaction with NCoR. The observation is particularly evident with wild-type receptors, which interact four to seven times better as TR-RXRα heterodimers with NCoRNID than with the isolated NCoR peptide displaying the highest affinity. Surprisingly, and in contrast to what was observed with short peptides, the mutations have little effect on the interaction with NCoRNID, which, however, remains very strong. The different behavior of mono- and heterodimeric TRs upon mutation is likely to reflect the role of RXRα that strengthens the recruitment of NCoR to the heterodimer, thereby masking the stabilizing effect of mutations. This hypothesis was confirmed by measuring and comparing the affinity of NCoRNID with wild-type and mutant TRβ monomers (Table 2 and Supplementary Fig. S6). As observed with the NCoR peptides, TRβ-Δ430 and TRβ-Δ432 alone displayed a higher affinity with the NCoRNID than TRβ.
Dissociation Constants (Kd in μM) Measured from Thermophoresis Experiments (MST) Between N-CoRNID Fragment and hRXRα–TR or hTR LBD Constructs in the Absence or Presence of Triiodothyronine
Together, these data show that pathologic mutations in TRα and TRβ induce a stronger interaction of mutant receptors with corepressors and a change in the mode of interaction of mutant receptors with corepressors by shifting the CoRNR binding specificity toward CoRNR1.
CoRNR1 β1 mediates increased corepressor recruitment by TR mutants
Having shown that the TRβ mutations strongly shift the CoRNR binding specificity toward SMRT (and to a lesser extend NCoR) CoRNR1, and that the amino-terminal region of CoRNR1 is involved in the interaction with wild-type TRs, the involvement of the CoRNR1 amino-terminal β1 strand in the interaction with mutant TRβ was assessed. First, shorter CoRNR1 sequences were used (N-CoRNR1 and S-CoRNR1 short; Fig. 1B), reducing the peptides to the strict helical motif LxxxIxxxIxxxF/Y. These shorter CoRNR1 peptides with deletion of their amino-terminal β1 region did not interact with TRβ-Δ432 in fluorescence anisotropy experiments, as was also observed for wild-type TRβ (Fig. 5).
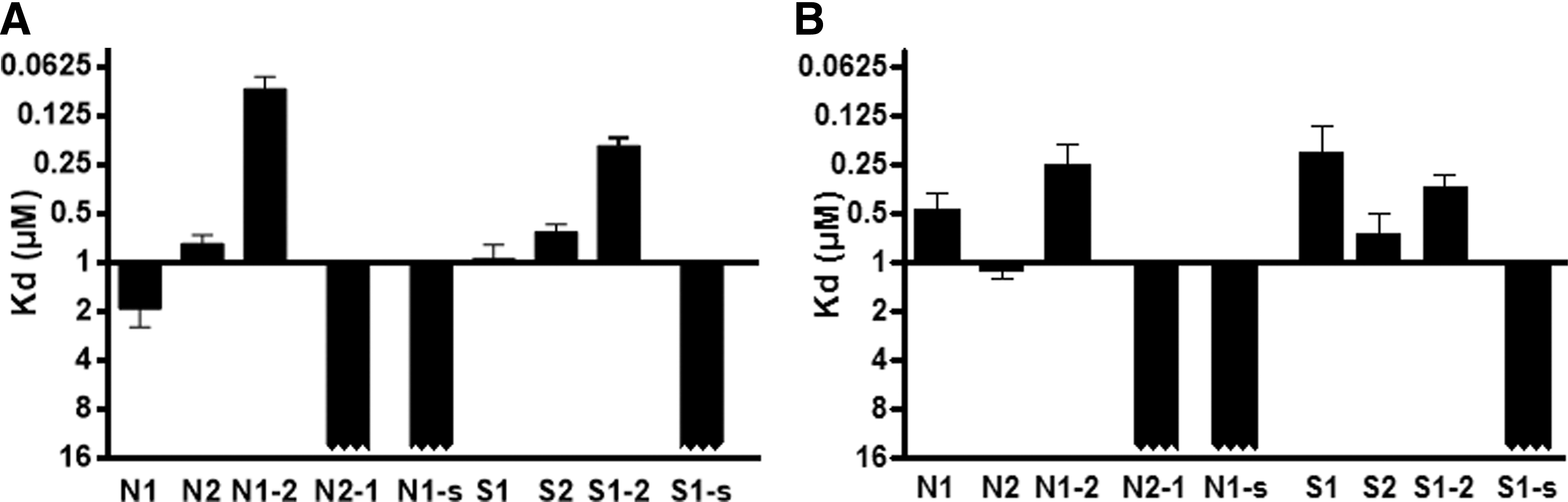
Importance of β1 strand of CoRNR1 in the binding of TRs to corepressors. Dissociation constants (Kd in μM) measured from fluorescence anisotropy titrations between fluorophore-labeled N-CoRNR1 (N1), N-CoRNR1-short (N1-s), N-CoRNR2 (N2), S-CoRNR1 (S1), S-CoRNR1-short (S1-s), S-CoRNR2 (S2), N-CoRNR1-2 (N1-2), N-CoRNR2-1 (N2-1), S-CoRNR1-2 (S1-2), and wild-type TRβ LBD (
Then, two types of chimeric peptides were used containing either the amino-terminal β1 region of CoRNR1 plus the consensus sequence from CoRNR2 (N-CoRNR1-2 and S-CoRNR1-2), or vice versa the amino-terminal region of NCoR CoRNR2 and the consensus sequence from CoRNR1 (N-CoRNR2-1; Fig. 1B). While the N-CoRNR2-1 chimera was unable to bind to TRβ and TRβ-Δ432, the N-CoRNR1-2 and S-CoRNR1-2 chimeric peptides displayed strong binding capacities, with Kds in the order of magnitude or even better than the native CoRNRs (Fig. 5).
Together, these experiments reveal that the amino-terminal β1 region of CoRNR1 from SMRT and NCoR plays a crucial role in the interaction of corepressors with wild-type and mutant TRβ. Hence, as mutant TRβs have an increased CoRNR1 recruitment, this was taken as an indication that the TRβ deletions could favor the β-sheet interaction between the TRβ variants and the corepressor CoRNR1 motif, similar to that seen for RAR and RevErb (15,16).
Mutations dramatically affect sensitivity to T3 agonist
In normal conditions, T3 acts as a natural agonist of TRs, inducing corepressor release and subsequent coactivator recruitment. First, the ability of T3 to dissociate the various CoRNR peptides and NCoRNID fragment from TRα and TRβ was measured. It was observed that all CoRNRs dissociate well from TRα and TRβ upon T3 addition (Fig. 2, Supplementary Fig. S1, and Table 1). For TRα-M388I, the efficiency of release of all N-CoRNRs was slightly lowered when compared to TRα, whereas a comparable efficiency was measured for the dissociation of S-CoRNRs (Fig. 2A and C and Table 1). Accordingly, NCoRNID dissociates only slightly less after addition of T3 from the RXRα-TRα-M388I heterodimer compared to wild-type RXRα-TRα (Table 2 and Supplementary Fig. S6).
In the case of the TRβ deletion mutants, great variability was observed in the T3-induced CoRNR release. It was noted that T3 can lower the interaction between both TR deletion mutants and some CoRNRs, which nevertheless retain a substantial affinity for the liganded receptors (e.g., TRβ-Δ430 and S- or N-CoRNR1; TRβ-Δ432 and S-CoRNR3). Whereas T3 efficiently releases S-CoRNR1, it fails to dissociate N-CoRNR1 from TRβ-Δ432 (Fig. 2E and Table 1). In line with this observation, NCoRNID could not be dissociated by T3 from the heterodimers containing either TRβ deletion mutant (Table 2 and Supplementary Fig. S6).
Using fluorescence anisotropy, the ability of native and mutant TRs to bind a fluorescein-labeled peptide containing the nuclear receptor box 2 (NR2) of the coactivator SRC1 in the absence or presence of T3 was then measured (Figs. 1B and 6 and Table 1). As expected, the affinity of unliganded TRs for SRC1-NR2 is poor (Kd ≥5 μM). Upon T3 addition, the binding affinity increases dramatically to around 0.3 μM for both wild-type receptor subtypes and TRα-M388I. This is in contrast with T3-bound TRβ-Δ432 that shows a very weak binding affinity for SRC1-NR2, and to a lesser extent T3-bound TRβ-Δ430.
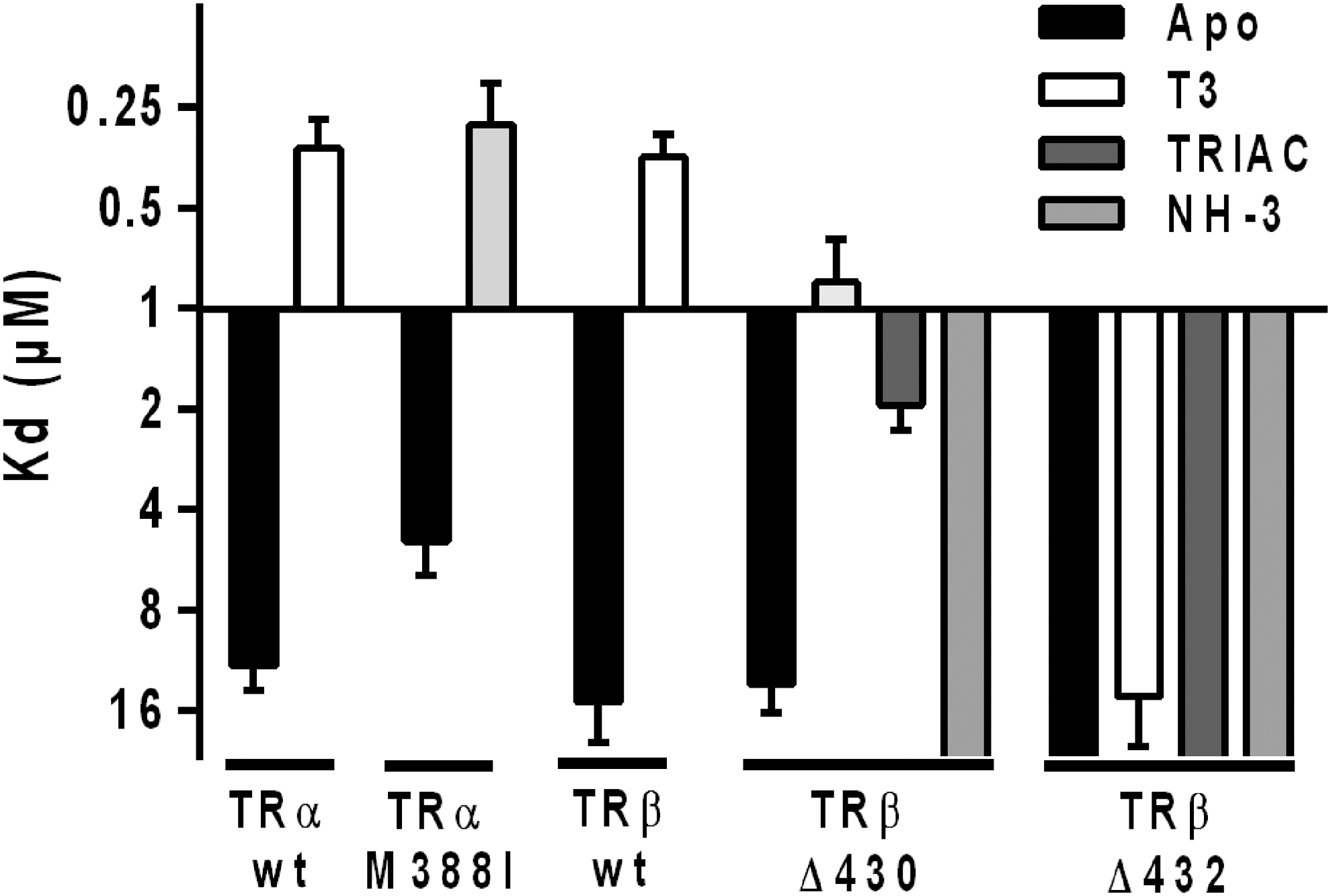
Affinities of wild-type and mutant TRs for a coactivator. Dissociation constants (Kd in μM) measured from fluorescence anisotropy titrations between fluorophore-labeled SRC1-NR2 and hTR-LBD constructs in the absence (black) or presence (white) of thyroid hormone T3. Affinities measured in the presence of TRIAC and NH-3 for both TRβ mutants are also shown.
Together, these data reveal that for TRβ mutants and to a lesser extent for the TRα mutant, T3 does not induce the complete release of all CoRNR motifs, some of them remaining attached to the ligand-bound receptors with high affinity. In addition, for TRβ-Δ432, T3 does not induce the binding of the coactivator peptide, whereas for TRβ-Δ430 T3 induces a reduced but measurable recruitment of the SRC1 peptide. Overall, a combination of decreased corepressor release and coactivator recruitment accounts for the graded insensitivity to thyroid hormone of these TR mutants.
Effect of TRIAC and NH-3 on corepressor and coactivator interaction with TRβ mutants
As the ligand T3 was not able to dissociate most of the corepressor peptides strongly from the TRβ mutants, the capability of other agonists of TRβ to induce the release of N-CoRNR1 peptide from TRβ-Δ430 and TRβ-Δ432 mutants was tested. Interestingly, whereas the agonist GC-1 was unable to dissociate N-CoRNR1, the agonist TRIAC efficiently decreased the affinity of the peptide for both TRβ-Δ430 and TRβ-Δ432 mutants (Table 3 and Supplementary Fig. S7). In fact, the Kd of the interactions between the N-CoRNR1 peptide and TRIAC-bound TRβ-Δ430 and TRβ-Δ432 decreased to 2.68 and 1.31 μM, respectively. Then, the study checked whether TRIAC was also able to dissociate the N-CoRNR2 peptide from both TRβ mutants and the NCORNID fragment from both RXRα/TRβ-Δ432 and RXRα/TRβ-430 heterodimers. As for N-CoRNR1, affinities of N-CoRNR2 for mutants strongly decreased in the presence of TRIAC (Table 3 and Supplementary Fig. S6). The affinity of NCORNID from both heterodimers was >40 times lower in the presence of TRIAC compared to the unliganded or T3-bound forms (Table 3 and Supplementary Fig. S7). In parallel, the same experiments were conducted with NH-3, a known TRβ antagonist. The ligand NH-3 strongly dissociated both N-CoRNR peptides from TRβ mutants (Table 3 and Supplementary Fig. S7) and prevented the interaction of the NCORNID fragment with both RXRα-TRβ-Δ432 and RXRα-TRβ-Δ430 heterodimers (Table 3 and Supplementary Fig. S6).
Dissociation Constants (Kd in μM) Measured from Fluorescence Anisotropy Titrations Between Fluorophore-Labeled NCoR (N-CoRNR1 and N-CoRNR2) and SRC1-NR2 Peptides and TRβ-Δ430 and TRβ-Δ432-LBD and from Thermophoresis Experiments Between N-CoRNID Fragment and RXRα/TRβ-Δ430 and RXR/TRβ-Δ432 LBD in the Absence or Presence of Different Ligands Agonists (T3, GC-1, and TRIAC) and Antagonist (NH-3)
Regarding the recruitment of the coactivator peptide (comprising the motif NR2 from SRC1), TRIAC exerted different effects on the two TRβ mutants. Similarly to T3-bound TRβ-Δ432, TRIAC-bound TRβΔ-432 displayed a very weak binding affinity for SRC1-NR2 (Table 3, Fig. 6, and Supplementary Fig. S7). On the contrary, TRIAC-bound TRβΔ-430 bound SRC1-NR2 with a reasonable affinity of about 2 μM (Table 3, Fig. 6, and Supplementary Fig. S7), slightly lower than for the T3-bound TRβ-Δ430. As expected, the antagonist NH-3 was not able to induce the recruitment of the coactivator peptide (SRC1-NR2) by both TRβ mutants (Table 3, Fig. 6, and Supplementary Fig. S7). Then, the effect of T3, TRIAC and NH-3 on the interaction of wild-type and mutant TRβ with coactivator was confirmed by a two-hybrid experiment with a construct harboring the nuclear interacting domain of human TIF2 (Supplementary Fig. S8A). Whereas wild-type TRβ efficiently recruits TIF2 in the presence of T3 and TRIAC at 1 and 10 μM, a modest but significant interaction between TRβ-Δ430 and the coactivator could be observed only at the highest ligand concentration, TRIAC being more effective than T3 at inducing a response. On the contrary, TRβ-Δ432 is not able to bind TIF2 in the presence of all the tested ligands.
Then, an experiment was carried out to show whether the agonist TRIAC was able to relieve the dominant-negative regulatory effect of the TRβ mutants more efficiently than T3. As shown in Supplementary Figure S8B, the unliganded TRβ-Δ430 and TRβ-Δ432 display an inhibitory action on wild-type TRβ, and TRIAC was slightly more potent than T3 at inducing a transcriptional response from both TRβ-Δ430 and TRβ-Δ432.
In conclusion, it was found that contrary to T3, TRIAC is an efficient inducer of corepressors dissociation from TRβ-Δ432 mutant. However, TRIAC was not able to induce the recruitment of coactivator by TRβ-Δ432, suggesting that this mutant cannot adopt a stable agonist conformation. Thus, it was hypothesized that TRIAC could bind or stabilize TRβ-Δ432 with higher affinity than T3, explaining the different effects induced by these ligands. In the case of the TRβ-Δ430 mutant, T3 did not induce the release of all CoRNR motifs, whereas TRIAC and NH-3 were quite potent in releasing all NCoR peptides and the NCORNID fragment from the RXRα/TRβ-Δ430 heterodimer. Moreover, both T3 and TRIAC were able to induce the recruitment of the coactivator by the protein. Although rather qualitative than quantitative, cell-based assays confirmed the general trend of the in vitro assays and revealed the better response of TRβ-Δ430 to TRIAC compared to T3 in two-hybrid and dominant-activity experiments.
TRβ mutants have lower binding affinities for T3 than for TRIAC and NH-3
Having shown different responses of T3, TRIAC, and NH-3 ligands on corepressor release and coactivator recruitment by the TRβ-Δ430 and TRβ-Δ432 mutants, the ability of TRα/β mutants to bind and to be stabilized by these ligands was investigated. First, thermal shift assays were used to compare the thermal stability of TRβ wild-type and mutants upon T3, TRIAC, and NH-3 binding. As expected, knowing the strong binding affinities of all three ligands, TRβ was strongly stabilized upon T3, TRIAC, and NH-3 binding, with a ΔT m of >7°C in the presence of only 0.5 equivalent of ligands (Fig. 7A). Of note, TRIAC induced a stronger stabilization of TRβ than T3 and NH-3 (Fig. 7A). For TRβ-Δ430, T3 did not induce any stabilization of the protein, even at 5 molar excess. In contrast, a slight but significant stabilization of the protein, with a ΔT m of 4.2°C and 3.2°C, was observed in the presence of 5 molar excess of TRIAC and NH-3, respectively. The stronger stabilization of TRβ-Δ430 by TRIAC and NH-3 when compared with T3 could be correlated with the differential effects of these ligands on corepressor release. The TRβ-Δ432 mutant in its apo form has a high T m of about 58°C, and T3 does not induce any stabilization of the protein, even at the highest ligand concentration. However, TRIAC and NH-3 were able to induce a slight stabilization of about 2.4°C and 3.3°C, respectively. These data suggest that TRIAC and NH-3 can bind to and stabilize TRβ-Δ430 and TRβ-Δ432, whereas T3, possibly due to a weaker binding affinity for TRβ mutants, is unable to do so.
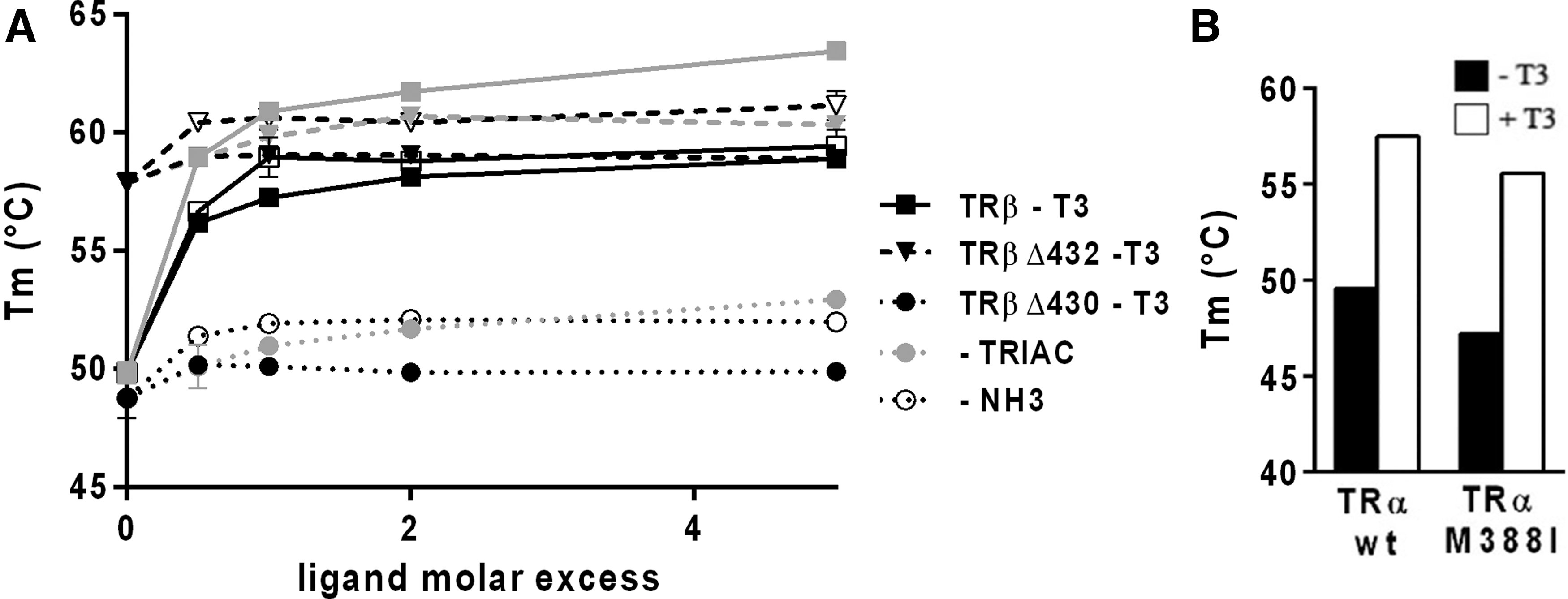
Effect of ligands on the thermal stability of wild-type and mutant TRs. (
Regarding TRα-M388I, only a slight decrease in the stability of the mutant could be observed compared to the wild-type receptor and a significant shift of the melting temperature induced by the addition of T3 (Fig. 7B). In agreement with the interaction studies reported above, these Thermofluor data suggest that the mutation M388I does not significantly affect the binding of hormone to TRα.
Second, a ligand titration assay was conducted to assess the relative potencies of the ligands in dissociating the N-CoRNR1 peptide from the TRβ-Δ430 and TRβ-Δ432 proteins. The potency of TR ligands reflects both their binding affinity for the receptor, as well as the affinity with which the N-CoRNR1 peptide is able to interact with the resulting ligand–TR complex. The fluorescence anisotropy binding curves for these ligand titrations show that all ligands induced a concentration-dependent dissociation of N-CoRNR1 from the TRβ mutants (Fig. 8). For the TRβ-Δ430 mutant, the IC50 values were 3.91 ± 1.92 and 2.28 ± 1.73 μM for TRIAC and NH-3, respectively, but >50 μM for T3. For TRβ-Δ432, IC50 values of 0.83 ± 0.72, 15.26 ± 5.30 μM, and >40 μM were measured for NH-3, TRIAC, and T3, respectively. As a comparison, an IC50 value of 0.16 ± 0.10 μM could be deduced from the titration of the N-CoRNR1–wt TRβ complex by T3, showing that T3 dissociated N-CoRNR1 peptide from wild-type TRβ at much more lower concentration (>250 times less) than for the TRβ mutants. Thus, these data validate the hypothesis that both mutants have lost their high affinity for the natural hormone T3, whereas NH-3 (TRβ-Δ430 and TRβ-Δ432), and to a lesser extent TRIAC (TRβ-Δ430), retain binding affinities in the micromolar range for the TR mutants. In conclusion, TR ligand potency in terms of N-CoRNR1 displacement follows the order: NH-3 > TRIAC > T3 for both TRβ mutants.
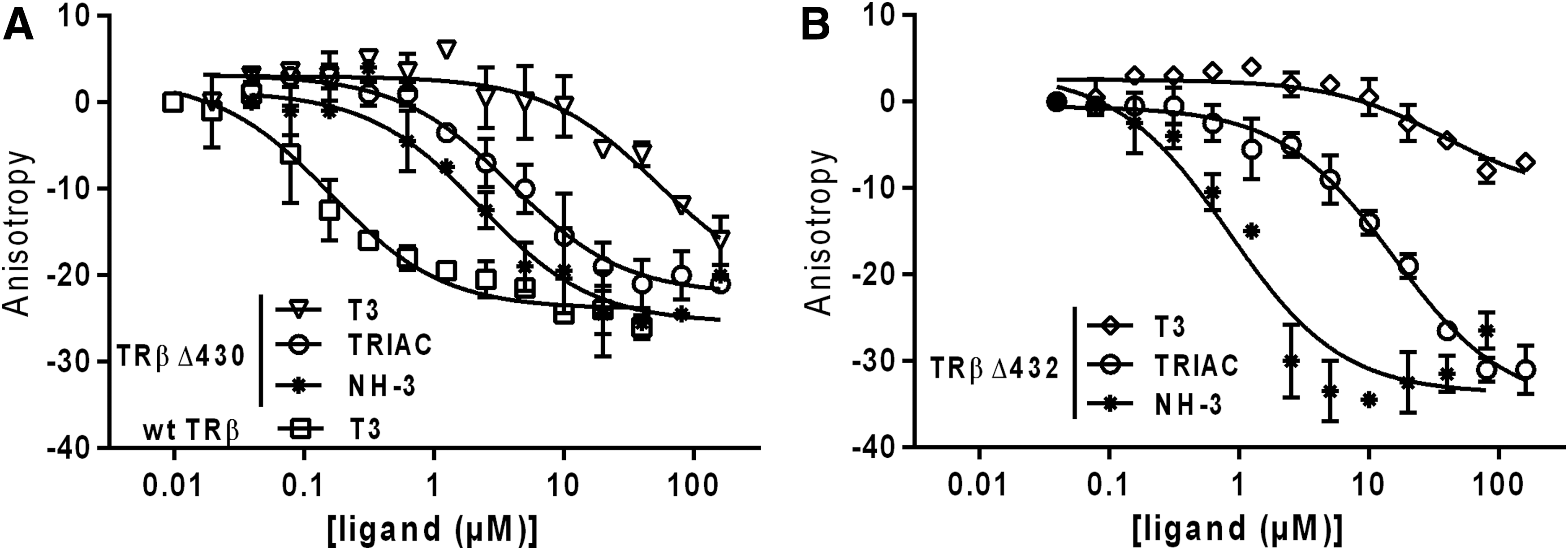
Ligand potency for N-CoRNR1 dissociation from TRβ mutants (ligand titration). (
All together, these results show that TRβ-Δ430 and TRβ-Δ432 mutants display a very weak affinity for T3, whereas TRIAC and NH-3 bind significantly to both mutants. These differences are likely to account for the differential effect of TRIAC, NH-3, and T3 on corepressor release.
Discussion
This study of pathological TRα M388I and TRβ deletion mutants reveals several important properties of the interactions between TRs and corepressors. First, the study confirms that both TR subtypes are able to bind the three CoRNR motifs of corepressors, even if they do so with different affinities and that both show preferential binding to SMRT boxes. Whereas it was initially shown that TRs interact with CoRNR3 of NCoR (40), this study shows that TRs interact more strongly with CoRNR3 of SMRT.
Contrary to what was already published (41), the study shows that the mutation M388I does not significantly modify the affinity of TRα for T3, as it has the same thermal stabilizing effect on the LBD of wild-type and mutant TRα. Indeed, in a native structure of chicken TRα in complex with T3 (PDB code 3UVV) (42), the equivalent methionine (M386) does not make any contact with the ligand T3. Furthermore, two crystal structures of TRα in complex with T3 were obtained with the M388 mutated into cysteine, and in these structures, T3 is still bound to the receptor (PDB codes 2H79 and 2H77) (43). Hence, the pathogenic effect of the M388I mutation has more to do with the global increase of affinity measured for both corepressors and coactivators. Even located at the carboxy-terminus of H11, this point mutation does not seem to induce drastic conformational changes of the receptor. Nevertheless, the M388 shows hydrophobic interactions with helix H12 in wild-type TRα (PDB code 3UVV) in order to maintain a favorable conformation for interaction with coregulators, and it is presumed that such contacts are lost in the M388I mutant, resulting in helix H12 destabilization.
A number of crystal structures of NR in complexes with CoRNR1 and CoRNR2 motifs (Table 1) have revealed the interaction surface of these CoRNR motifs with NRs. For CoRNR1 interaction, the β-strand of CoRNR1 interacts with S3 in place of H11 of NR LBD, and its carboxy-terminal helix docks into the coregulator binding cleft of NR LBD, as described for RAR and RevErb (15,16). For CoRNR2 interaction, the position of helixes H11 and H12 of NR LBD defines a hydrophobic surface comprising the carboxy-terminal part of helix H3, helix H4, and the loop linking them to accommodate the helical motifs of CoRNR2. Using NMR, this study shows that interaction of TRs with CoRNR2 and CoRNR1 presumably also follows this paradigm. Moreover, the first structural characterization of the CoRNR3 motif was obtained, isolated and in complex with TRs. CoRNR3 folds as a transient helix all along the motif that mediates the interaction with TRs, and the STD effects measured on the complex are similar to those observed for the TR/CoRNR2 complex. Thus, as the three different binding mechanisms occur in TRs, it is suggested that TRs are able to undergo different conformations in the H11–H12 region. Accordingly, Mengeling et al. showed that both the helical and the β-strand portions of the CoRNR1 are important for the interaction of the peptide with TRα (44).
Interestingly, the pathological TRβ mutations studied here are all located in the dynamic helix H11 region and cause defects in TR activity. This study shows that the TRβ-Δ430 and TRβ-Δ432 mutants display a stronger affinity for corepressors than the wild-type TRβ and a transfer of specificity toward CoRNR1. Notably, previous studies using other methods have yielded results consistent with these conclusions as to the importance of CoRNR1 and its β-strand in the TR interaction (20,22,44,45). It is suggested that the point deletions affect the structure of TRβ by promoting the dynamic equilibrium between H11 and S3 to be displaced toward S3, and/or by increasing the flexible departure of H12 from the corepressor interaction surface. This conformational change locks the TRβ mutants in a repressive state, explaining why they function as dominant-negative receptors (45). Furthermore, this study shows that TRβ mutations affect T3 binding, since no hormone-induced thermal stabilization could be measured in the presence of this ligand, contrary to wild-type TRβ, and the study estimated a low potency of T3 for both TRβ mutants by ligand titration in corepressor dissociation assays. These data are in line with previous results showing that these TRβ-Δ430 and TRβ-Δ432 mutants fail to bind T3 hormone at a detectable level (45). However, in these mutants, T3 still lowers the dissociation constant of some CoRNR peptides (e.g., N-CoRNR3 and S-CoRNR1), and it is presumed that the binding of T3 is not completely impaired by these mutations.
The thyroid hormone analogue TRIAC and NH-3 are a known agonist and antagonist of TRs, respectively, and they were shown to interact strongly with TRβ (46,47). This study shows that TRIAC and NH-3 induce a significant thermal stabilization of TRβ-Δ430 and TRβ-Δ432 mutants, suggesting that they bind to these TRβ mutants with a higher affinity than T3. The study then confirmed by ligand titration in corepressor dissociation assays that in contrast to T3, NH-3 and TRIAC retain a significant binding affinity for both TRβ mutants. Thus, the deletions Δ430 and Δ432 appear to influence ligand binding by either disrupting direct contacts with the ligand or altering interactions that stabilize the bound conformation. However, the fact that these mutations are each a deletion of one residue renders the modeling of the TRβ mutants, as well as the prediction of the effect of these deletions on ligand binding, difficult and even impossible. Crystal structures of wild-type TRβ in complex with T3 or TRIAC have indeed revealed a crucial role of residues from helix H11 in ligand binding (PDB code 1XZX (48) and PDB code 3JZC (46,49)). In particular, the histidine 435 represents an important site of contact with both T3 and TRIAC (Supplementary Fig. S9). In the TRβ-Δ430 and TRβ-Δ432 mutants, this residue may no longer be correctly positioned to maintain this stabilizing contact with T3 and TRIAC. On the other side, TRIAC and T3 are differently accommodated by side chain flexibility in the hydrophilic portion of the LBD (Supplementary Fig. S9). Whereas T3 is in direct contact with R282, the charged and polar substituent at position 1 of the inner ring of TRIAC makes contact with R320 and, in this case, the R282 side chain rotates and forms a hydrogen bound with N331.
Then, very interestingly, it was observed that TRIAC is able to dissociate corepressor peptides from TRβ-Δ430 and TRβ-Δ432 mutants and a NCORNID fragment from the corresponding heterodimers strongly. This observation could have valuable therapeutic applications by reducing the abnormal repression caused by TRβ-Δ430 and TRβ-Δ432 mutants. A recent publication reports that TRIAC was successfully used for a treatment of a patient with the TRβ-Δ430 mutant (50), in line with the ability of TRIAC to induce the recruitment of coactivator to TRβ-Δ430. On the contrary, the TRβ-Δ432 mutant does not seem able to adopt an agonist conformation, even in presence of TRIAC, as no recruitment of coactivator is induced in this case. It is hypothesized that TRIAC will induce only a de-repression of TRβ-Δ432 but not a full activation, probably due to an inability of this receptor to adopt an active conformation. Thus, the deletions Δ430 and Δ432 seem to vary in their effect on ligand binding and also in the ability of the TRIAC to convert the mutated receptor to the agonist conformation. On the other hand, NH-3 is even more efficient than TRIAC at dissociating corepressors from both TRβ deletion mutants. However, as already observed for wild-type TRβ (47), NH-3 is unable to promote interactions between TRβ-Δ430 and TRβ-Δ432, and the coactivator peptide that is likely one of the underlying mechanisms for its observed antagonist activity.
The in vitro characterization of the recruitment of coregulators by wild-type and TRβ mutants, and of the differential effects of ligands on these interactions, provides a clear molecular understanding of the pathogenicity of these mutations and highlights the pharmacologic potential of TRIAC. Extension of the results to cell-based experiments provided supporting but somewhat attenuated effects compared to those observed in vitro. Nevertheless, it is highlighted that new interfaces between TR and corepressors are generated by the three studied mutated TRs involved in the development of RCCC and RTH. The data provide a rational explanation for the dominant-negative behavior of these mutants, resulting from an increased affinity of corepressor for all mutants. This enhanced interaction with SMRT (and to a lesser extent NCoR), and enhanced dominant-negative/repression properties has been further demonstrated to be a common property of mutations that, similar to Δ430 and Δ432, affect helix 11/12 of the TRs (20,22,51 –54). Intriguingly a possible correlation between these TR mutants that enhance SMRT binding and the pituitary form of the RTH syndrome has been speculated on in the literature (55). In addition, the changes in the affinity of TR mutants for corepressors as assayed here with the TR ligand binding domains and in the absence of DNA have also been generally observed in assays that do incorporate DNA (20,22,44,45,56,57). Nonetheless, the potential of DNA recognition to alter coregulator recruitment by the TRs and other NRs further is an important question that will require further studies. In addition, the study demonstrates an impairment of T3 binding by TRβ-Δ430 and TRβ-Δ432 LBDs compared to wild-type TRβ to be a cause of the inefficiency of T3 to release corepressors from mutant TRβs. Very interestingly, it was found that contrary to T3, the agonist TRIAC and the antagonist NH-3 are able to disrupt abnormal interactions between corepressors and pathological TRβ-Δ430 and TRβ-Δ432, giving rational evidence that TRIAC could play a role in the treatment of syndromes linked to mutations in the carboxy-terminal region of the LBD of TRβ.
Footnotes
Acknowledgments
The CBS is a member of the France-BioImaging (FBI) and the French Infrastructure for Integrated Structural Biology (FRISBI), two national infrastructures supported by the French National Research Agency (ANR-10-INBS-04-01 and ANR-10-INBS-05, respectively). We acknowledge the platforms of the Grenoble Instruct center (ISBG; UMS 3518 CNRS-CEA-UGA-EMBL) supported by the French Infrastructure for Integrated Structural Biology Initiative FRISBI (ANR-10-INSB-05-02) and by the Grenoble Alliance for Integrated Structural Cell Biology GRAL (ANR-10-LABX-49-01) within the Grenoble Partnership for Structural Biology (PSB). We acknowledge the Laboratory of Spectroscopy and Calorimetry (LEC) at Brazilian Biosciences National Laboratory (LNBio), CNPEM, Campinas, Brazil for their support with the use of equipment (Monolith NT.115 Microscale Thermophoresis device, NanoTemper Technologies GmbH). We acknowledge Taisa Ribeiro Ferreira (LNBio) for technical support. We acknowledge the financial support from the CNPq Programa Ciencia Sem Fronteiras (BJT 300143/2015-0 to ALM), from the FAPESP (Fapesp grant # 2016/15986-7 to EV), from the Association pour la Recherche contre le Cancer (ARC grant to DH) and from the TGIR-RMN-THC Fr3050 CNRS (HD).
Author Disclosure Statement
The authors have nothing to declare.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.