
Abstract
Background:
Dual oxidases (DUOX1 and DUOX2) were initially identified as H2O2 sources involved in thyroid hormone synthesis. Congenital hypothyroidism (CH) resulting from inactivating mutations in the DUOX2 gene highlighted that DUOX2 is the major H2O2 provider to thyroperoxidase. The role of DUOX1 in the thyroid remains unknown. A recent study suggests that it could compensate for DUOX2 deficiency in CH. Both DUOX enzymes and their respective maturation factors DUOXA1 and DUOXA2 form a stable complex at the cell surface, which is fundamental for their enzymatic activity. Recently, intra- and intermolecular disulfide bridges were identified that are essential for the structure and the function of the DUOX2–DUOXA2 complex. This study investigated the involvement of cysteine residues conserved in DUOX1 toward the formation of disulfide bridges, which could be important for the function of the DUOX1DUOXA1 complex.
Methods:
To analyze the role of these cysteine residues in both the targeting and function of dual oxidase, different human DUOX1 mutants were constructed, where the cysteine residues were replaced with glycine. The effect of these mutations on cell surface expression and H2O2-generating activity of the DUOX1–DUOXA1 complex was analyzed.
Results:
Mutations of two cysteine residues (C118 and C1165), involved in the formation of the intramolecular disulfide bridge between the N-terminal ectodomain and one of the extracellular loops, mildly altered the function and the targeting of DUOX1, while this bridge is crucial for DUOX2 function. Unlike DUOXA2, with respect to DUOX2, the stability of the maturation factor DUOXA1 is not dependent on the oxidative folding of DUOX1. Only mutation of C579 induced a strong alteration of both targeting and function of the oxidase by preventing the covalent interaction between DUOX1 and DUOXA1.
Conclusion:
An intermolecular disulfide bridge rather than an intramolecular disulfide bridge is important for both the trafficking and H2O2-generating activity of the DUOX1–DUOXA1 complex.
Introduction
D
DUOX1/2 belong to the NADPH oxidase (NOX/DUOX) family. In addition to their five counterparts (Nox1–5), which contain six transmembrane (TM) domains, the two DUOXs have an extended N-terminal extracellular domain called the peroxidase homology (PoxH) domain, followed by an additional TM segment and an intracellular loop containing two calcium-binding EF-hand motifs (1,2).
Each DUOX requires maturation factors (DUOXA1 and DUOXA2) to exit from the endoplasmic reticulum (ER) and reach the plasma membrane where they form a stable complex, which is a prerequisite for their H2O2-producing activity (6 –8). The discovery of biallelic mutations in DUOXA2 in patients with dyshormonogenesis further reinforces the role of the DUOX2–DUOXA2 complex in thyroid hormone biosynthesis (9).
DUOX enzymes are expressed as fully glycosylated forms at the plasma membrane (10). They produce O2 ·– as an intermediate product and H2O2 as a final product (11). The extra-cytosolic “A-loop” region of DUOXs, particularly in DUOX1, is an important structural determinant that interacts with the N-terminal of their partners (DUOXAs) and facilitates the conversion of O2 ·– to H2O2, possibly through mechanisms promoting the stabilization and maturation of DUOX–DUOXA complexes (12).
Despite their high level of sequence homology, the DUOXs present differences in their regulation at both the post-translational and transcriptional levels. Different phosphorylation pathways regulate the enzymes, with DUOX1 being activated by protein kinase A (Gs-PKA pathway), while DUOX2 activation occurs through protein kinase C (Gq-phospholipase C [PLC] pathway) (13). These enzymes are not restricted to the thyroid; they are also well expressed in the respiratory tract epithelium and in the gastrointestinal mucosa, where they play a key role in innate immunity (14 –16). Overexpression of each DUOX gene has been selectively associated with an increasing number of diseases such as pancreatic cancer (DUOX2) (17) and radio-induced tumors (DUOX1) (18), indicating a specific role, which is possibly related to their individual structures.
Recently, an intramolecular disulfide bridge was identified that is essential for the structure and function of DUOX2 (19). This disulfide bridge, formed in the ER, appears to be a key event in the trafficking of the DUOX2–DUOXA2 complex.
Since a study performed on the purified N-terminal peroxidase-like ectodomain of the counterpart DUOX1 also supports the existence of cysteine-mediated disulfide bridges for intermolecular protein–protein interactions (20), the effect of mutations of cysteine residues in this domain on the targeting of and extracellular H2O2 production by DUOX1 were analyzed.
Materials and Methods
Cell culture, mutagenesis, and transfection
HEK293 cells were cultured in Dulbecco's modified Eagle's medium (high glucose; PAA Laboratories) supplemented with 10% (v/v) fetal calf serum and penicillin/streptomycin (100 μg/mL; Life Technologies Ltd.). The mutagenesis of cysteine residues of DUOX2 has been previously performed using the QuikChange site-directed mutagenesis kit (Stratagene) and a pcDNA3 plasmid containing the full-length human (h) wild type (WT) DUOX2 (hDUOX2-pcDNA3) as a template (19,21). The NH2-terminal hemagglutinin epitope-tagged human DUOX1 (HA-DUOX1 pcDNA3) was kindly provided by Dr. Xavier de Deken (Université libre de Bruxelles, Brussels, Belgium), and the NH2-terminal V5 epitope-tagged human DUOXA1 (V5-DUOXA1-pcDNA3) and the COOH-terminal V5 epitope-tagged human DUOX1 (DUOXA1-V5-pcDNA3) were kindly provided by Dr. Thomas Leto (NIAID/NIH, Bethesda, MD) and were described elsewhere (7). The mutagenic primers used for cysteine mutation in DUOX1 are listed in Supplementary Table S1 (Supplementary Data are available online at
In transient cell transfection experiments, HEK293 cells reaching 50–70% confluence were transfected on 24-well plates using X-tremeGENE HP DNA Transfection Reagent according to the protocol recommended by the manufacturer (Roche).
Measurement of extracellular H2O2 generation in intact cells
H2O2 generation was quantified by the Amplex red/horseradish peroxidase assay (Sigma–Aldrich), which detects the accumulation of a fluorescent oxidized product, as previously described (19). The HA tag does not interfere in the extracellular H2O2-generating activity or the targeting of DUOXs (7). The presence of the c-myc tag in the C-terminal region of DUOXA2 does not alter the H2O2 generation of DUOX2 (12,19). Moreover, neither N-terminal nor C-terminal V5-tagged DUOXA1 affected the H2O2 generation of DUOX1 (Supplementary Fig. S1A and B).
Measurement of extracellular TPO activity in intact cells
Extracellular TPO enzymatic activity was determined from the stable human TPO-HEK293-expressing cell line transiently co-transfected with either both HA-DUOX1 (WT or mutant HA-DUOX1 C118G) and V5-DUOXA1 or either both HA-DUOX2 (WT or mutant C124G) and cMyc-DUOXA2. The enzymatic activity was monitored by measuring the oxidation of Amplex red by TPO in the presence of one of the two H2O2-generating systems (DUOX1/DUOXA1 or DUOX2/DUOXA2), as previously described (21).
Western blot analysis
The cell extracts were solubilized in denaturing sample buffer (Tris/HCl 10 nM, pH 7, 2.5% sodium dodecyl sulfate (SDS), 1 mM of EDTA, 1 mM of EGTA, 4 M of urea, 20 mM of N-ethylmaleimide, and protease and phosphatase inhibitor cocktails (Roche). The samples were then sonicated for 10 s. The protein samples (10–20 μg) supplemented with or without 0.1 M of dithiothreitol DTT (Sigma–Aldrich) and 10% glycerol were denatured for 5 min at 95°C, subjected to SDS polyacrylamide gel electrophoresis using an 8% or 10% Tris-glycine polyacrylamide gel and electrotransferred to 0.2 μm of Protran BA83 nitrocellulose sheets (Schleicher & Schuell). The immunodetection of DUOX was performed using serum from immunized rabbit clone 3679 (1:10,000 for DUOX2 and 1:1000 for DUOX1). Additionally, primary antibodies were used for immunodetection: anti-HA (1:500; clone 3F10; Santa Cruz Biotechnology), anti-cMyc (1:2000; Santa Cruz Biotechnology), anti-vinculin (1:5000; Abcam), and mouse monoclonal anti-V5 (1:15,000; Thermo Fisher Scientific). The blots were subsequently incubated with horseradish peroxidate–conjugated or alkaline phosphatase–conjugated secondary antibodies for 1 h.
Immunoprecipitation
After transfection, the proteins were immunoprecipitated (IP) using HA antibody, as recommended by the kit manufacturer (Sigma–Aldrich).
Flow immunocytofluorometry
For surface protein staining, the cells were detached from the plates with phosphate-buffered saline (PBS) containing 5 mM of EDTA/EGTA and incubated respectively for 30 min at room temperature with a rat anti-HA of high affinity (clone 3F10; Roche) or with a mouse monoclonal anti-V5 (Thermo Fisher Scientific) for NH2-terminal HA epitope-tagged human DUOX1 and DUOX2 and NH2-terminal V5 epitope-tagged human DUOXA1 surface immunofluorescence staining. The samples were washed once with D-PBS/0.1% bovine serum albumin (BSA) and incubated for 30 min with Alexa 488-conjugated anti-rat IgG or Alexa 568-conjugated anti-mouse immunoglobulin G (IgG) diluted in D-PBS/0.1% BSA (Life Technologies Ltd.). After two washing steps, the cells were re-suspended in D-PBS/0.1% BSA. Fluorescence was assayed using a BD Accuri™ C6 Flow Cytometer (BD Biosciences), counting 10,000 events per sample. Relative protein expression was determined by calculating the differences in total fluorescence between the sample and an equal-sized population of pcDNA3-DUOX1 or DUOX2 and pcDNA3-DUOXA1 or DUOXA2-transfected cells corresponding to 100%.
Immunofluorescence studies
Transfected cells grown on glass coverslips were washed in D-PBS and fixed in 4% paraformaldehyde/D-PBS for 10 min. Nonspecific binding sites were blocked with 3% BSA/D-PBS for 30 min. For staining of NH2-terminal HA epitope-tagged human DUOX1 or NH2-terminal V5 epitope-tagged human DUOXA1, the cells were incubated for 2 h with rat anti-HA (1:100) clone 3F10 or with mouse monoclonal anti-V5 (1:400; Thermo Fisher Scientific) in Tris-buffered saline (TBS) containing 0.3% BSA. After three washes in TBS with 0.1% Tween, Alexa 488-conjugated anti-rat IgG or Alexa 568-conjugated anti-mouse IgG diluted (1:200) in TBS/0.3% BSA (Life Technologies Ltd.) was used as the secondary antibody. After two washes in TBS 0.1% Tween and staining with DAPI fluorochrome (Life Technologies Ltd.), the slides were mounted with Dako Fluorescence Mounting Medium (Dako Denmark). Fluorescent images were captured on a confocal microscope (Leica DM IRE 2; Perkin Elmer).
Statistical analysis
Data are represented as the mean ± standard deviations of the results of at least three independent experiments. Student's t-test was used to calculate the significance of values.
Results
Existence of an intramolecular disulfide bridge affecting in part the activity and targeting of DUOX1
The predicted amino acid sequences of the two DUOX paralogs (DUOX1: 1551 amino acids; DUOX2: 1548 amino acids) share 83% sequence similarity. Comparison between the primary structure of the DUOX1 and DUOX2 enzymes highlights that several cysteines are highly conserved phylogenetically for both enzymes, suggesting a key role of these residues in the structure and function of these enzymes (Supplementary Fig. S2). Recently, an intramolecular disulfide bridge between C124 and C1162, residing respectively in the N-terminal ectodomain and in an extracellular loop connecting TMs 4 and 5 of DUOX2, was identified, which is essential for the structure and enzymatic function (19). The C124 corresponds to C118 and C1162 to C1165 in DUOX1 (Fig. 1A and B). To analyze the role of these cysteine residues in the targeting and function of DUOX1, two human HA-DUOX1 mutants were constructed, where both cysteine residues were replaced with glycine. Unlike what was highlighted for DUOX2, these mutations significantly decrease but do not totally suppress cell surface expression, as analyzed by fluorescence-activated cell sorting and immunofluorescence (Fig. 1C and D), and H2O2-generating activity of DUOX1 (Fig. 1E). It has been shown that the intramolecular disulfide bridge C124–C1162, formed in the oxidized state, leads to modification of electrophoretic mobility of DUOX2 in non-reducing conditions. As observed with DUOX2, under non-reducing conditions, the electrophoretic mobility of the WT DUOX1 decreased; DUOX1 was detected at approximately 250 kDa instead of at 185 kDa when evaluated under reducing conditions. Mutation of cysteines 118 and 1165 suppressed the mobility shift of DUOX1, indicating that these two cysteines are additionally involved in the oxidative folding of DUOX1 and form an intramolecular disulfide bridge (Fig. 2A). However, unlike DUOX2, the absence of oxidative folding in mutant DUOX1 mildly altered the function and the targeting of the enzyme. Thus, the formation of the intramolecular disulfide bridge between the N-terminal ectodomain and one of the extracellular loops, which is crucial for DUOX2 function, is less important for DUOX1.
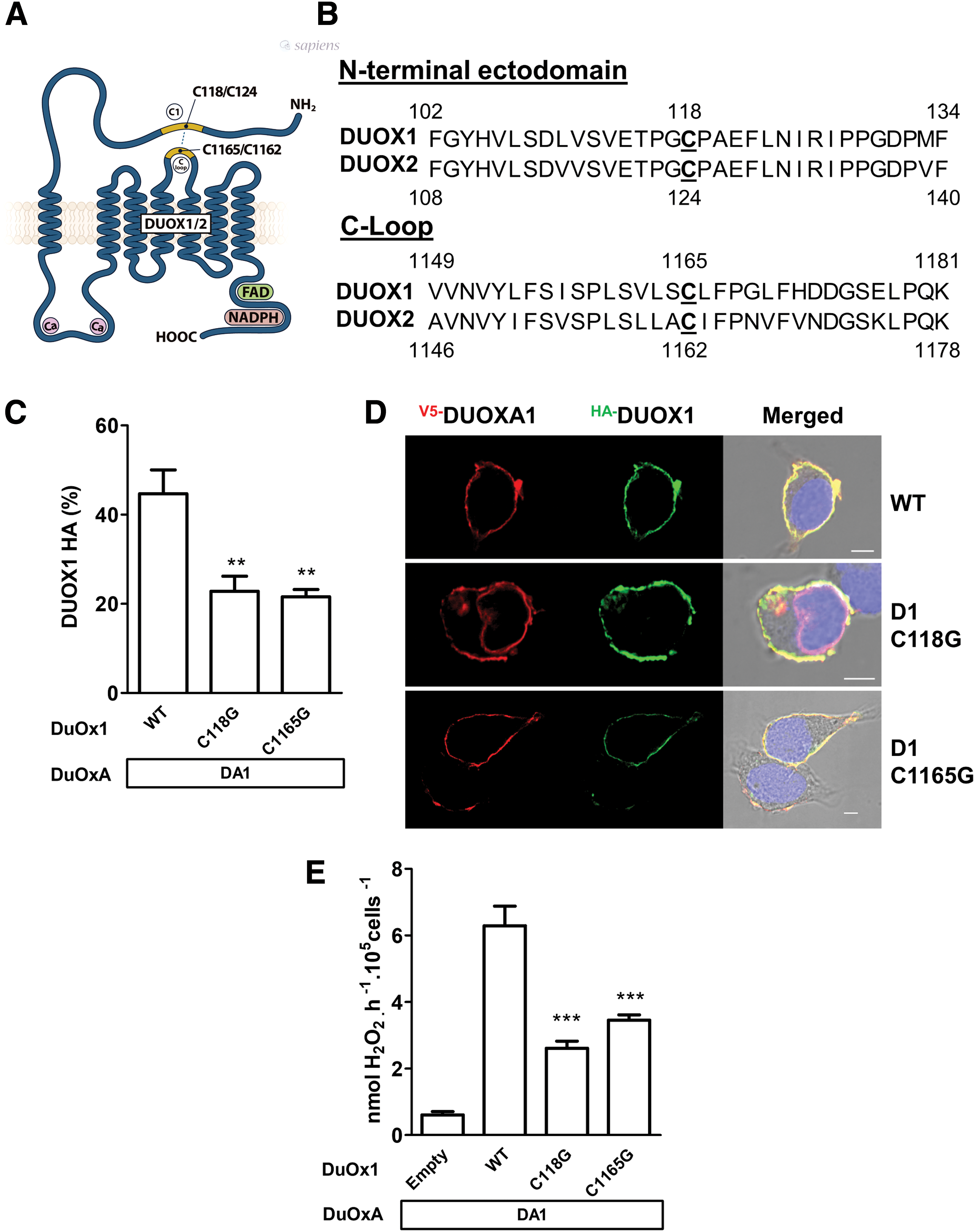
The intramolecular disulfide bridge between the first cysteine in the N-terminal domain and C-loop domain is not required for plasma membrane targeting and H2O2 production of DUOX1. (

Differently of DUOX2, DUOX1 mutant could provide sufficient quantities of H2O2 for TPO activity. (
DUOX2 is the major provider of H2O2 for TPO for thyroid hormone biosynthesis. Since DUOX1 is suggested to compensate for DUOX2 deficiency, the ability of the DUOXs and their respective mutants to make TPO active at the cell surface were compared (Fig. 2B). Both WT DUOXs have accomplished this function. By contrast, only DUOX1 mutant could provide sufficient quantities of H2O2 for TPO activity (Fig. 2B).
Each maturation factor DUOXA is essential and specific for reconstitution of DUOX-based oxidases in vitro. However, functional studies have suggested that DUOXA1 can replace DUOXA2 with DUOX2. Here, it was observed that even if both DUOXAs were able to target DUOX2 correctly at the cell membrane (Fig. 3A), the extracellular H2O2-generating activity remained totally dependent on DUOXA2 and was barely measurable in the presence of DUOXA1 (Fig. 3B). Mutation of C124 (C1) abolished both targeting and function of DUOX2, irrespective of the maturation factor used. As previously shown, this was associated with a total absence of the electrophoretic mobility shift of DUOX2 observed under non-reducing conditions (Fig. 3C). However, the results obtained with DUOX1 were quite different. Independent of the complex formed with each of the DUOXAs, the mutation of the first cysteine decreased but did not abolish the targeting of DUOX1 at the plasma membrane (Fig. 3D). However, the activity of mutant DUOX1 remained totally dependent on its own maturation factor (Fig. 3E). In addition, unlike what was seen with the mutant DUOX2 C124G (C1; Fig. 3C), the nature of the partner modified the electrophoretic migration of mutant DUOX1 C118G (C1) under non-reducing conditions to a greater or lesser extent, which appeared somewhat delayed when co-expressed with DUOXA1 (Fig. 3F). This was associated with detection of an additional band very close in size, approximately 180 kDa, and corresponding to a glycosylated state, which is in agreement with expression at the cell surface. Therefore, even if DUOXA2 was able to promote a partial targeting of DUOX1 and, to a lesser extent, that of mutant DUOX1 C1, it could not promote post-translational modification of DUOX1 such as glycosylation, which is a prerequisite for its activity at the cell surface.
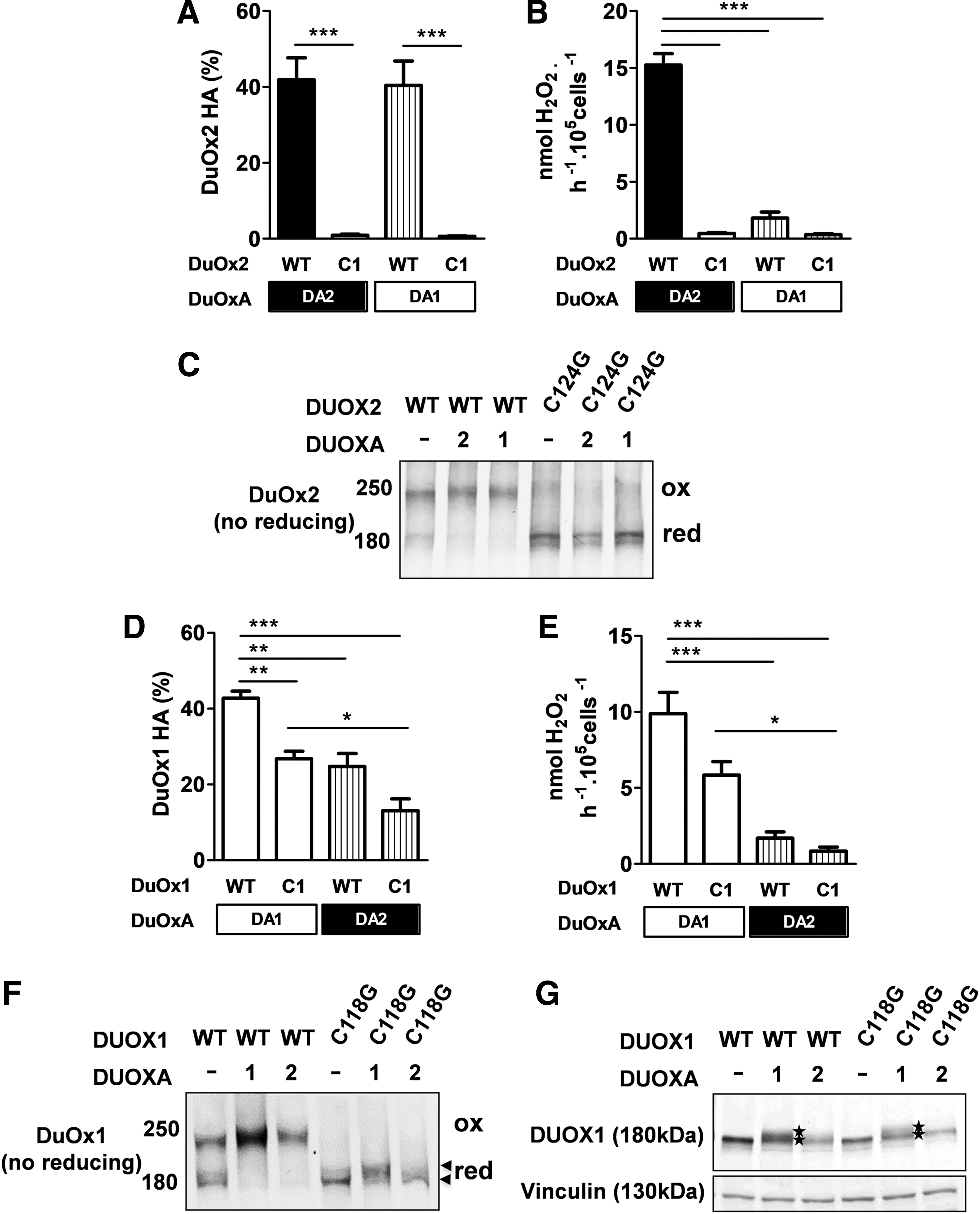
DUOX function remains completely dependent on its own maturation factor. (
Unlike DUOXA2, with respect to DUOX2, the stability of the maturation factor DUOXA1 is not dependent on the oxidative folding of DUOX1
It was previously shown that a correct structural conformation of DUOX2 is essential for the stability of DUOXA2 (19). Interestingly, the level of expression of DUOXA2 was decreased when it was co-expressed with WT DUOX1, and this decrease was more pronounced with mutant DUOX1 C118G (Fig. 4A). This indicates that the stability of DUOXA2 is highly specific for DUOX2. By contrast, the maturation factor DUOXA1 appeared to be stable, regardless of the mutation state of DUOX expressed, indicating that the stability of each maturation factor is differently impacted by the oxidized state of both DUOXs (Fig. 4B).
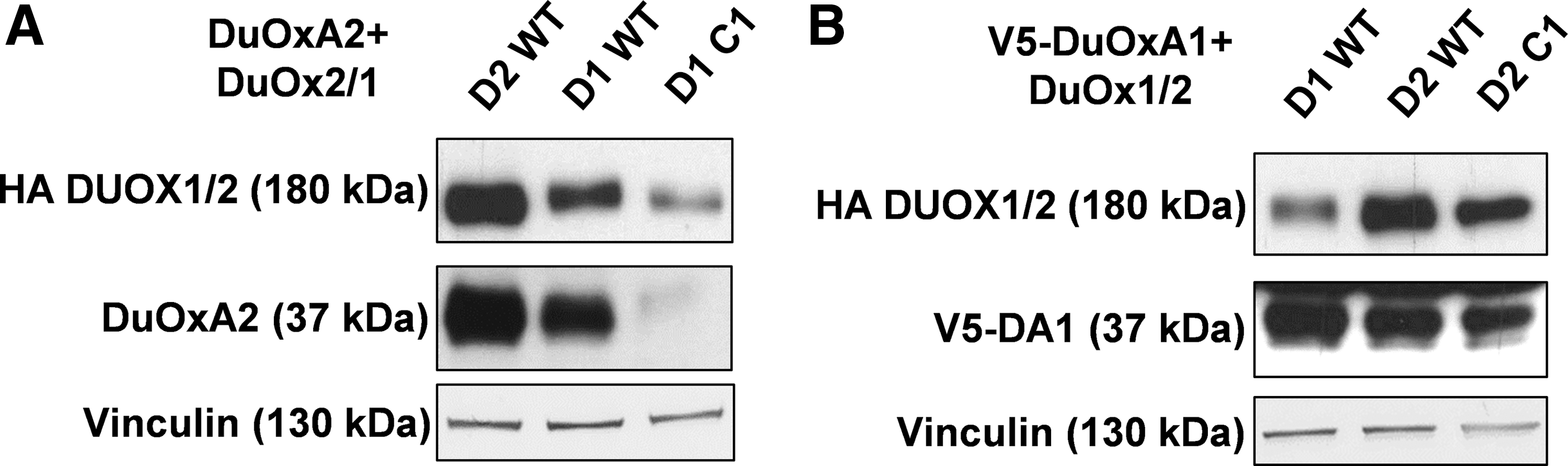
The stability of each maturation factor is differently impacted by the oxidative folding of DUOXs. The expression of DUOX1/2 and DUOXA1/2 was analyzed by Western blot. (
Of note, immunoblot analysis revealed that DUOXA1 and DUOX1 are stabilized by each other (Fig. 5A). When they are expressed separately in the cells, their level of expression is lower, suggesting degradation via the proteasome-dependent pathway. Treatment of DUOXA1-tranfected HEK293 cells with MG132, a proteasome inhibitor, enhanced the expression of DUOXA1 in a dose-dependent manner. Recently, we showed that DUOX2 interacts covalently with DUOXA2 through an intermolecular disulfide bridge, which is also important for the stability of DUOXA2, and as expected, DUOX2 immunoprecipitated with DUOXA2. Immunoprecipitation experiments confirmed that even if DUOXA1 was well expressed in the presence of DUOX2, the interaction between both proteins was very weak (Fig. 5B) (7). There was also a strong interaction between DUOX1 and its maturation factor DUOXA1. Although DUOXA2 was partly stabilized in the presence of DUOX1, the interaction between the two proteins was non-existent. The strong interaction between the DUOX enzymes and their respective DUOXA was correlated with a H2O2-generating activity measured in particulate membrane fractions, which contained the plasma membrane and all intracellular membrane compartments, particularly the ER, where both proteins are synthesized and chiefly expressed. Thus, as previously shown (7), despite a high homology between the DUOX enzymes and their respective maturation factor DUOXA, the specific interactions between each other govern, in the early step of the maturation, the functional conformation of each H2O2-generating system.

The interactions between DUOXs and their maturation factor DUOXAs appear very specific. (
Unlike DUOX2, the oxidative folding of DUOX1 is not a key event in trafficking and reactive oxygen species production
As conducted previously for DUOX2, the role of the five cysteine residues residing in the N-terminal ectodomain of DUOX1 in both the targeting and function of NADPH oxidase was analyzed. The cysteine residues (118, 345, 364, 565, and 579), which are all conserved in DUOX1 of different species and in DUOX2, were replaced with glycine (Fig. 6A and B and Supplementary Fig. S2). As previously observed (20), only mutation of C579 totally abolished extracellular activity. This was associated with suppression of the electrophoretic mobility shift under non-reducing conditions, indicating that this cysteine plays a critical role in correct structural conformation of DUOX1 related to the enzymatic function. Mutation of other cysteine residues led to a mild decrease in H2O2 generation (Fig. 6C). In addition, the two cysteine mutations (C118G and C579G), which impact the oxidative folding of DUOX1, did not affect the expression of DUOXA1, indicating that the stability of DUOXA1 does not depend on the N-terminal conformation of DUOX1.
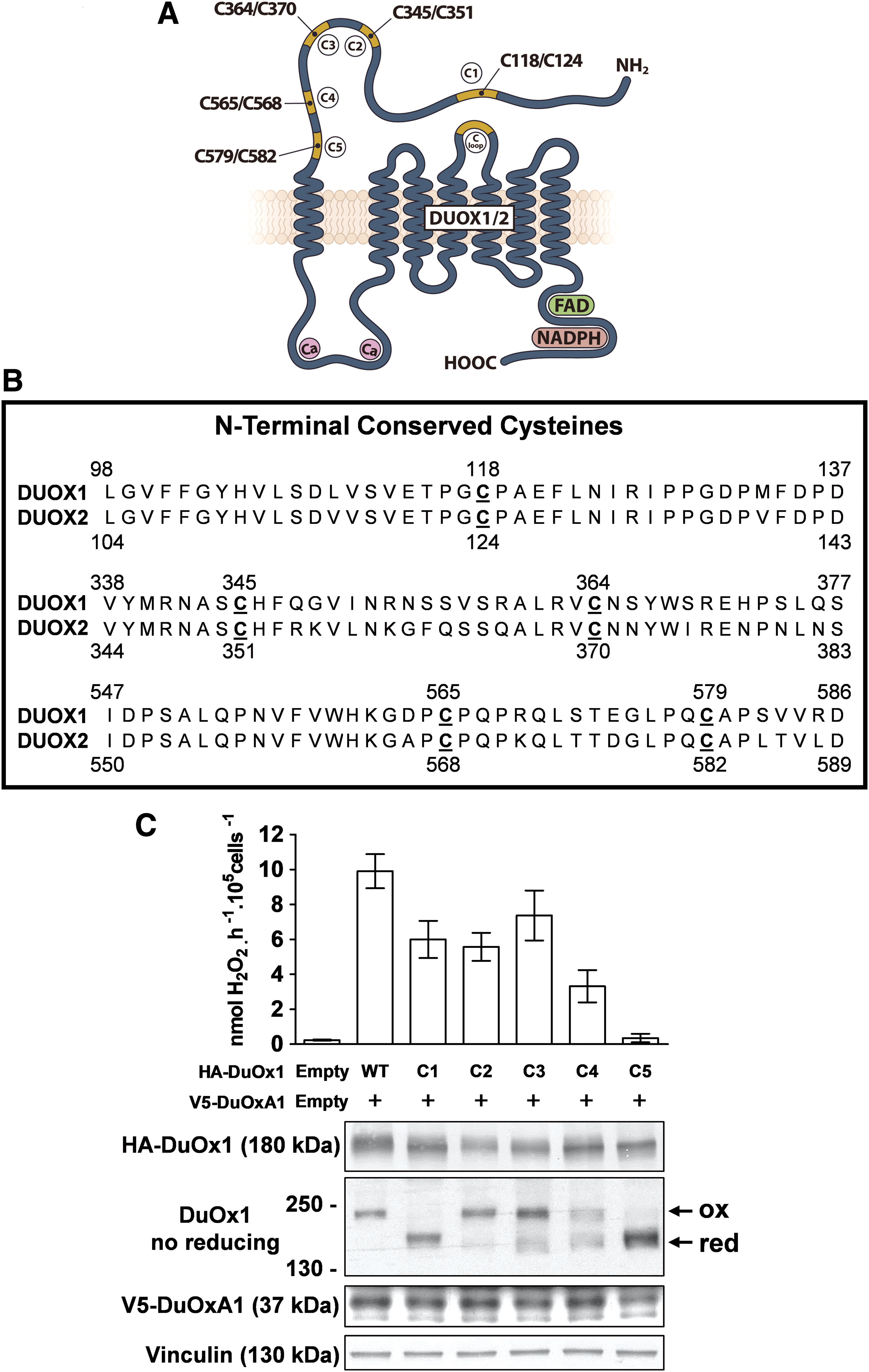
Effects of cysteine mutations in the N-terminal domain of DUOX1 on its H2O2-generating activity and on the stability of DUOXA1. (
As observed with whole cells, only H2O2 generation of particulate fractions from mutant C579G DUOX1-transfected cells was completely abolished (Supplementary S3). Additionally, this mutation completely prevented targeting of the complex DUOX1/DUOXA1 at the cell surface (Fig. 7A and B), which appeared condensed in the perinuclear zone (Fig. 7C). Immunoprecipitation experiments showed that mutation of cysteines impacted the interaction between DUOX1 and DUOXA1 at different levels, with a weaker detectable interaction observed when mutant C565 and C579 of DUOX1 were co-expressed with DUOXA1. These two cysteines, conserved in DUOX2, were recently shown to be involved in intermolecular disulfide bridges between DUOX2 and DUOXA2, which are important for the function of the complex (19). Regarding DUOX1, only mutation of C579 promoted a strong alteration of both targeting and function of the oxidase. Moreover, this mutation caused an absence of maturation of DUOX1, indicating that the covalent interaction between DUOX1 and DUOXA1, involving this cysteine, is a prerequisite for its maturation and therefore for its functional expression at the cell surface (Fig. 7D and E). In addition, comparison of the electrophoretic migration of mutant DUOX1 C118G and C579G under non-reducing conditions underlined that despite the absence of a major shift, which is related to the absence of oxidative folding of DUOX1, there was a slight delayed migration for mutant C118, highlighting a difference in the maturation process between the two mutants (Fig. 7F). Unlike DUOX2 mutant, its counterpart DUOX1 C118G is able to associate with DUOXA1, exit from the RE, and reach the plasma membrane. Thus, the oxidative folding has less of an impact on DUOX1 targeting and function.
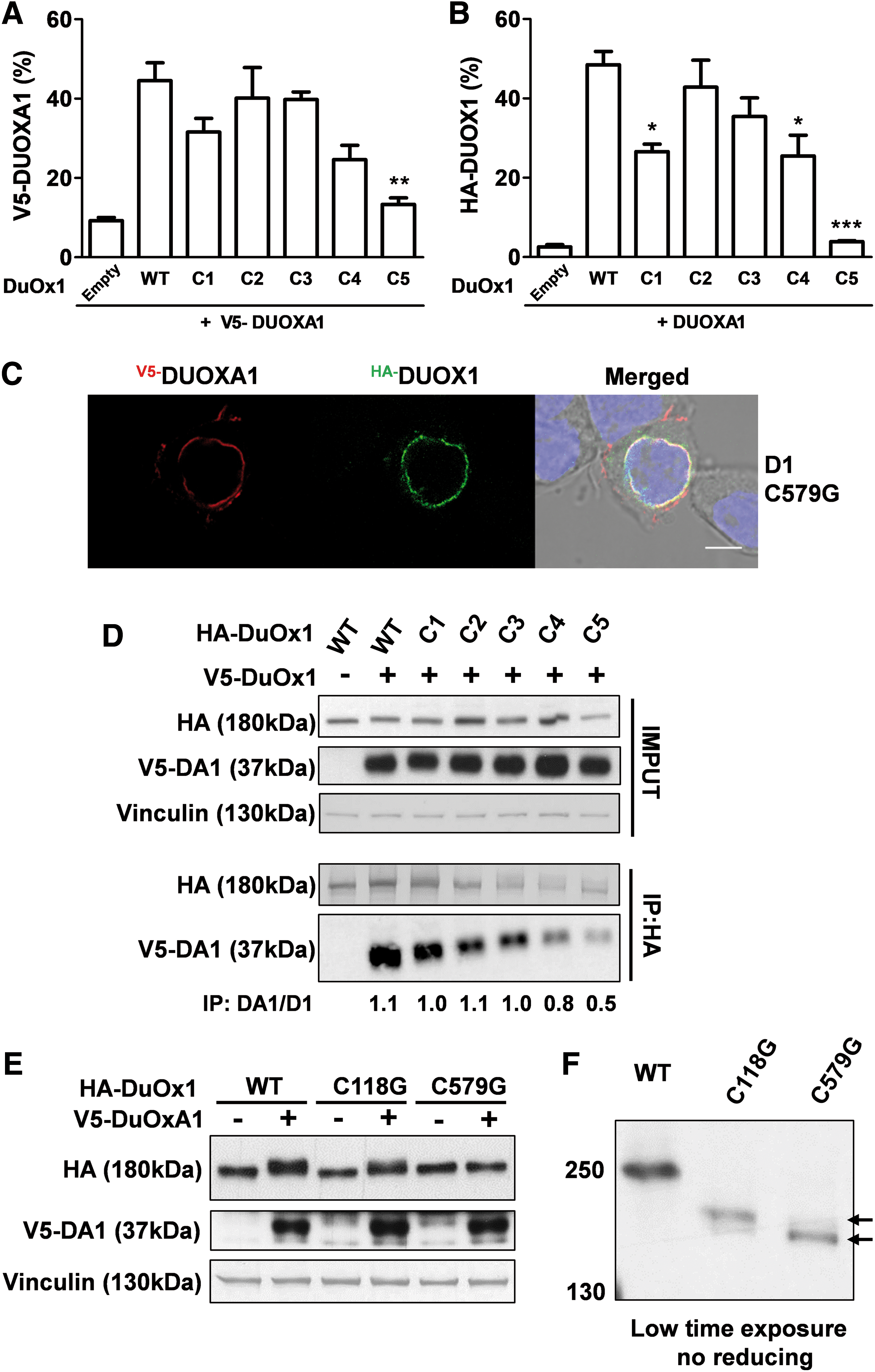
Only the C579G mutation strongly affects both DUOX1 targeting and function. (
Discussion
Recently, it was shown that mutations of cysteine residues in the N-terminal domain of the human DUOX2 protein affect both targeting and extracellular H2O2 production of the enzyme, indicating that these residues are critical for its proper maturation (19). In particular, the existence of an intramolecular disulfide bridge between the N-terminal domain and the second predicted extracellular loop of DUOX2 involving the cysteine residues C124 and C1162, respectively, was described. This disulfide bridge has an important structural implication in both export and activity of DUOX2 by promoting the covalent interaction between DUOX2 and its functional partner DUOXA2. Moreover, this interaction is critical for DUOXA2 stability. Although DUOX1 shares 83% sequence similarity with DUOX2, mutation of the cysteine residues of the N-terminal domain of DUOX1, which are conserved between DUOXs, had different effects on the stability, function, and partner interaction of DUOX1. In particular, this study reveals that the intramolecular disulfide bridge between the cysteine residues 118 and 1165 is not a key event in the trafficking of the DUOX1/DUOXA1 complex. Suppression of this bridge mildly affected both targeting to the plasma membrane and the extracellular H2O2-generating activity of DUOX1. As observed with DUOXA2, DUOXA1 is stabilized when it is co-expressed with DUOX1. However, unlike DUOXA2, whose stability and covalent interaction are strictly dependent on the oxidative folding of DUOX2, DUOXA1 could be stabilized by DUOX2. Despite the absence of a strong interaction, this result was illustrated by immunoprecipitation experiments. This stability is related to the cell surface expression of the DUOXs but not necessarily related to their activity, which remains totally dependent on their respective maturation factors. Thus, the export function of DUOXA can be dissociated from that exerted on the DUOX activity.
Interestingly, beyond its role as a maturation factor for DUOX1, DUOXA1 may also have other independent functional roles. Indeed, it has been shown to interact with p53, with which it is implicated in neuronal cell differentiation (22). This could also explain why the stability of DUOXA1 is not strictly dependent on DUOX1.
This study additionally confirmed that among all the mutated cysteine residues, only C579 completely abolished both the targeting and activity of DUOX1 (20). Unlike that observed for DUOX2, this mutation totally suppressed the shift in the electrophoretic mobility under non-reducing conditions, indicating the importance of this cysteine in a conformation-dependent mobility shift. In addition, this mutation strongly impacts the interaction with DUOXA1, which is a prerequisite for the functional maturation of DUOX1. Unlike that observed for the DUOXA2/DUOX2 complex, both the stability and function of DUOXA1 are not dependent on the oxidative folding of DUOX1. It appears that it is an intermolecular disulfide bridge, and not an intramolecular disulfide bridge, that is the first key event in the trafficking of the DUOX1/DUOXA1 complex.
The results are summarized in Figure 8, which highlights the major differences between DUOX1 and DUOX2. Interestingly, several DUOX2 mutations have been reported in the ectodomain and the extracellular loop, in close proximity to the cysteine residues whose redox regulation has functional importance (19), in patients with CH (23 –27). These mutations may confer structural changes in both domains and thus prevent disulfide bridge formation, which is a prerequisite for enzymatic function. Recently, it was demonstrated that a missense mutation G201E reported in patients with CH causes the suppression of a shift in the electrophoretic mobility of mutant DUOX2 under non-reducing conditions, corroborating this model (28). The data show that the DUOX1–DUOXA1 complex can provide H2O2 for thyroperoxidase to be active at the cell surface. Therefore, it can be envisaged that DUOX1 could compensate the dysfunction of DUOX2, particularly in adult life, when the thyroid hormone requirements are decreased.
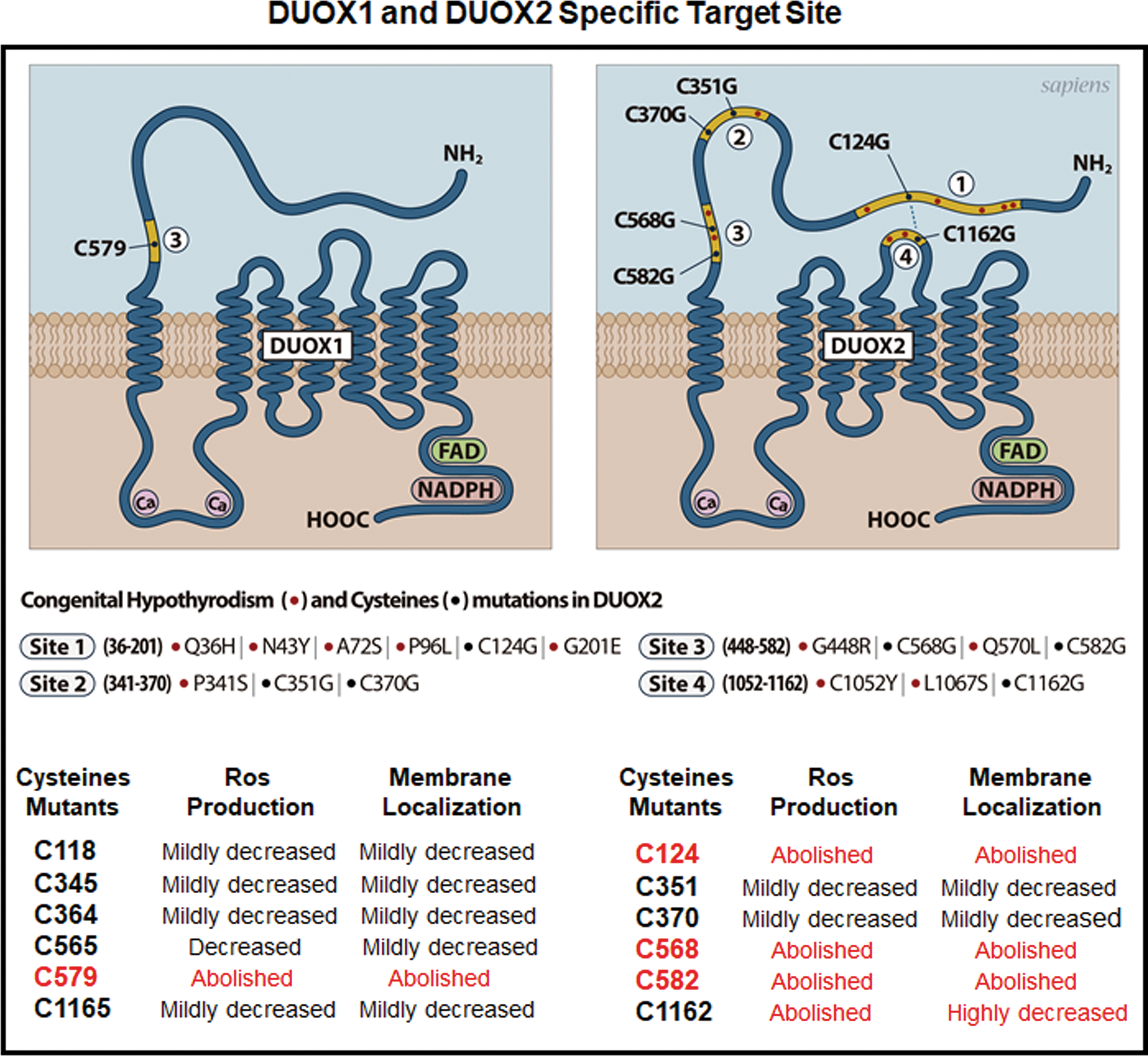
Proposed schematic representations of DUOX1 and DUOX2-identified mutation sites. The N-terminal domain presents key sites involved in both oxidative folding and function of DUOX1 and DUOX2. Several mutations identified in patients with congenital hypothyroidism are present in the N-terminal domain of DUOX2. The yellow line underlines the regions whose mutations significantly impair H2O2 production. Within such regions, cysteines that are mutated in DUOX2 (19) and in DUOX1 (in this article) are indicated. Compared to DUOX2, only one mutation of cysteines in the N-terminal domain of DUOX1 abolishes both H2O2 production and surface expression. Color images available online at
Except for a DUOX1 mutation resulting in aberrant splicing and a protein truncation, no naturally occurring DUOX1 functional mutations have yet been described in patients with thyroid dysfunction. The role of DUOX1 in the biology of this organ is unknown. Although digenic mutations in DUOX1 and DUOX2 are associated with severe CH, suggesting that DUOX1 may compensate for the DUOX2 deficiency in some patients with transient CH (5), other roles for this enzyme are possibly yet to be determined. DUOX1-dependent H2O2 generation might modulate redox-dependent signaling that controls differentiation, cytoskeletal arrangement, and/or cell migration. Recently, ionizing radiation, a strong risk factor for the development of thyroid cancer, was determined to cause strong induction and activation of DUOX1 in thyrocytes and induce genomic instability by DUOX1-derived H2O2 (18). Therefore, deciphering the structural differences between DUOX1 and DUOX2 will aid us in designing therapeutics targeting these enzymes to control their reactive oxygen species–generating activity that could be deleterious in pathological conditions.
Footnotes
Acknowledgments
We gratefully acknowledge Dr. H. Gasberger for the DUOXA2-expressing vector, Dr. X. de Deken for the DUOX1-expressing vector, and Dr. T. Leto for the DUOXA1-expressing vector. This work was supported by grants from Electricité de France (EDF) and Ligue Contre le Cancer (comité du Val-de Marne). R.A.L. is a recipient of a fellowship from Institut National Du Cancer (INCA). F.H. and J.C. are both recipients of fellowships from Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, Brazil). C.B. is a recipient of a fellowship from Fondation de France.
Author Disclosure Statement
No competing financial interests exist.