
Abstract
Background:
The prognosis of advanced or metastatic medullary thyroid carcinoma (MTC) is poor, and there are few therapeutic options. Anlotinib has previously shown promising antitumor activity on MTC in preclinical models and a Phase I study. This Phase II clinical trial was devised to confirm the antitumor activity of anlotinib in patients with advanced or metastatic MTC.
Methods:
Patients with unresectable locally advanced or metastatic MTC received once daily oral anlotinib 12 mg, two weeks on/one week off, until disease progression, death, unacceptable toxicity, or withdrawal of consent for any reason. The dose was adjusted on the basis of observed toxicity. The primary endpoint was progression-free survival (PFS).
Results:
Fifty-eight patients received anlotinib treatment. The primary endpoint PFS has not yet been reached at the time of analysis. On the basis of investigator assessments, 56.9% of patients experienced a partial response. PFS rate at 48 weeks was 85.5%. Forty-five patients had a ≥50% decrease in serum calcitonin concentration from baseline. The most common adverse events were hand-foot syndrome, hypertriglyceridemia, cholesterol elevation, fatigue, and proteinuria.
Conclusions:
Anlotinib demonstrated a durable antitumor activity with a manageable adverse event profile in locally advanced or metastatic MTC.
Introduction
M
The only way to cure MTC is the complete resection of thyroid tumor and any locoregional metastases (2,8). For progressive or symptomatic metastatic MTC that cannot be treated with local management, systemic therapy should be considered. However, traditional cytotoxic agents, such as 5-fluorouracil, doxorubicin, dacarbazine, and cyclophosphamide, showed limited efficacy but significant toxicity (2,9,10).
One of the validated therapeutic targets for MTC is the RET oncogene (11,12), which is mutated in the germline in almost all of the patients with hereditary MTC (6,13), and 50–60% of the patients with sporadic MTC harbor somatic RET gene mutations (14,15). Besides, angiogenesis plays a crucial role in the growth and dissemination of thyroid cancer cells. Vascular endothelial growth factor receptors (VEGFR-1 and VEGFR-2) (16,17), as well as fibroblast growth factor/fibroblast growth factor receptor (FGFR), are frequently overexpressed in MTC tumor cells and vascular endothelium (18 –20).
Vandetanib and cabozantinib, small molecular inhibitors that target RET and VEGFR-2, provide therapeutic efficacy in MTC by blocking both angiogenic and proliferative pathways (3,21 –23). Both vandetanib and cabozantinib have been approved by the Food and Drug Administration (FDA) for treating MTC. Unfortunately, neither has been approved for MTC treatment in China. Many other tyrosine kinase inhibitors such as lenvatinib, motesanib, and axitinib (among others) have shown various degrees of activity in Phase II clinical trials (24 –26).
Anlotinib is a novel tyrosine kinase inhibitor targeting multiple receptor kinases involved in tumor proliferation, vasculature, and the tumor microenvironment (27). Anlotinib inhibits VEGF/VEGFR signaling by selectively targeting VEGFR2/3 and FGFR1–4 with high affinity. Anlotinib also suppresses the activity of platelet-derived growth factor receptor α/β, c-Kit, and RET, showing significant inhibition of tumor proliferation (27). In preclinical experiments, anlotinib showed broad antitumor activity against a variety of xenograft models (27,28).
In a Phase I study, anlotinib showed manageable toxicity and broad-spectrum antitumor potential. Anlotinib at doses from 5 to 16 mg was administered to patients with solid tumors once a day in two schedules: four consecutive weeks (4/0) or two weeks on/one week off (2/1) (27). Dose-limiting toxicity was grade 3 hypertension at 10 mg in the 4/0 schedule, and grade 3 hypertension and grade 3 fatigue at 16 mg in the 2/1 schedule. Pharmacokinetic assessment indicated that the half-life of anlotinib at 12 mg in the 2/1 schedule was 116 hours. The maximum tolerated dose, 12 mg once daily in the 2/1 schedule, was chosen for expanding the study. The main serious adverse effects were hypertension, hypertriglyceridemia, hand-foot syndrome, and increased lipase levels (27).
Based on these promising results, this Phase II study was performed to assess the antitumor effect of anlotinib in patients with locally advanced or metastatic MTC. Moreover, the tolerability of anlotinib in MTC was also evaluated.
Methods
Study design and participants
In this single-arm Phase II study, patients were enrolled at eight institutions across China. This study was conducted in accordance with the principles of the Declaration of Helsinki and the Good Clinical Practice guidelines of the International Council for Harmonisation. All patients provided written informed consent to participate in the study. Pathology materials (tumor blocks or representative slides) were centrally evaluated.
Key eligibility criteria included age between 18 and 70 years, histologically confirmed unresectable or metastatic MTC, at least one measurable lesion by computed tomography scan according to Response Evaluation Criteria In Solid Tumors (RECIST) v1.1, Eastern Cooperative Oncology Group (ECOG) performance status 0–2, life expectancy of at least three months, calcitonin serum levels ≥500 pg/mL, and adequate bone marrow, renal, hepatic, and cardiac function checked within seven days before the start of the study drug, as evidenced by the following: (i) absolute neutrophil count ≥1500/mm3, (ii) platelets ≥80,000/mm3, (iii) hemoglobin ≥10.0 g/dL (without blood transfusion within 14 days), (iv) serum creatinine ≤1.0 × upper limit of normal (ULN), (v) bilirubin ≤1.25 × ULN, (vi) aspartate aminotransferase and alanine aminotransferase ≤1.5 × ULN (≤5.0 × ULN for patients with liver involvement), (vii) fasting triglycerides ≤3.0 mmol/L, (viii) fasting cholesterol ≤7.75 mmol/L, and (ix) Doppler ultrasound confirmed left ventricular ejection fraction ≥50%. Women of childbearing potential and male patients had to agree to use adequate contraception during the study participation and up to six months following completion of therapy.
Patients were excluded if they met any of the following criteria at the time of screening: had received a prior treatment of anti-angiogenic agents, including sunitinib, sorafenib, bevacizumab, imatinib, and apatinib; reported a previous or concomitant malignancy, which probably affects life expectancy, except curative skin basal cell carcinoma and cervical carcinoma in situ; received chemotherapy or radiotherapy within 28 days before the start of initiation of the study therapy; participated in other clinical trial within 28 days before the start of initiation of the study therapy; had an ongoing toxicity greater than grade 1 according to the National Cancer Institute's Common Terminology Criteria for Adverse Events v4.0 (CTCAE v4.0); unable to swallow oral medications; any malabsorption condition; a known history of brain or meningeal metastasis and spinal compression; diagnosed with severe or uncontrolled disease, including symptomatic congestive heart failure, unstable angina (angina symptoms at rest), or myocardial infarction, within six months before day 1 of treatment; severe or uncontrolled cardiac arrhythmias; uncontrolled hypertension (systolic pressure >140 mmHg or diastolic pressure >90 mmHg); active or uncontrolled infection; known history of liver cirrhosis, decompensated liver diseases, and chronic active hepatitis; poorly controlled diabetes (fasting blood glucose >10 mmol/L); spot urine with 2+ or more protein and a 24-hour urine collection with a total protein excretion >1000 mg/24 hours; a non-healing wound, ulcer, or bone fracture; evidence or history of disease with bleeding potential or therapeutically treated with an anticoagulant agent such as warfarin, heparin, or analogue; arterial or venous thrombotic or embolic events such as cerebrovascular accident (including transient ischemic attacks), deep-vein thrombosis, or pulmonary embolism; known history of human immunodeficiency or organ allograft; or lactation or women with a positive pregnancy test in blood or urine within seven days before initiation of the study therapy.
The trial was approved by the Institutional Review Board of each participating institution and conducted in accordance with guidelines for Good Clinical Practice and the Declaration of Helsinki. All patients provided written informed consent. The trial was registered at clinicaltrials.gov (registration number: NCT01874873).
Procedures
Eligible patients received oral anlotinib 12 mg once daily, two weeks on/one week off, until disease progression (RECIST v1.1 guidelines), death, unacceptable toxicity, or withdrawal of consent for any reasons. During the treatment period, assessments of the tumor status were performed every six weeks.
Dose modifications for adverse events (AEs) were done according to the protocol. Clinical assessments of safety, including medical history, physical examination, and laboratory tests, were done every three weeks during the first 24 weeks and then at six-week intervals thereafter. AEs were graded according to National Cancer Institute's CTCAE v4.0. All patients were followed until death from any cause or withdrawal of consent. The primary endpoint was progression-free survival (PFS) according to RECIST v1.1. Secondary endpoints were objective response rate (ORR), disease control rate (DCR) at 24 weeks, OS, biochemical response (change of serum calcitonin levels from baseline), and safety.
Statistical analysis
DCR at 24 weeks associated with best supportive therapy and anlotinib in patients with locally advanced or metastatic MTC was determined as 50% and 70%, respectively. A Simon two-stage testing procedure was applied with type I error of 5% and type II error of 20% each (α = 0.05, β = 0.2). On the basis of optimal design principle, 15 patients were enrolled in the first stage. If 8/15 patients did not have disease progression at 24 weeks, 28 patients were further enrolled in the second stage. If 26/43 patients did not show disease progression at 24 weeks, the result would be positive. A surplus recruitment of five patients was allowed to correct for ineligible or untreated patients. In this study, another 10 eligible patients were enrolled after obtaining approval from the Institutional Review Board.
PFS was defined as the time from treatment initiation to the date of disease progression or death from any cause, whichever came first. Patients alive at the time of analysis were censored at the date of last disease assessment. OS was measured from the date of treatment initiation to the date of death (from any cause). PFS and OS were estimated using Kaplan–Meier survival curves. ORR was defined as the proportion of patients who had a partial or complete response. DCR was defined as the proportion of patients who had achieved complete response, partial response, or stable disease at 24 weeks.
The following patient populations were considered in the final analyses: full analysis set (FAS)—all patients who were eligible and had started their allocated treatment (at least one dose of the study drug); per protocol set (PPS)—all patients who were eligible and received their allocated treatment for at least six weeks; and safety set (SS)—all patients who had started treatment (at least one dose of the study drug).
Results
Between July 2013 and July 2014, 58 eligible patients were recruited to this study. The final data analysis was carried out in July 2016. Table 1 contains the baseline characteristics of the patients. The median age was 46.9 years (range 22–71 years). A total of 93.1% of the patients underwent surgery, 25.9% received radiotherapy, and 12.1% received chemotherapy before study entry. A total of 89.7% of the patients were diagnosed with metastatic MTC. The most common metastatic sites were the lymph nodes, lungs, and liver.
ECOG PS, Eastern Cooperative Oncology Group performance status.
All patients started treatment according to protocol. Three patients were excluded from the PPS because of a lack of target lesions (n = 2) or withdrawal from the study within one week (n = 1). Therefore, 58 patients were included in the FAS and the SS, and 55 patients were included in the PPS. All 58 patients were treated, receiving a total of 908 cycles (median 16; range 1–30).
Efficacy
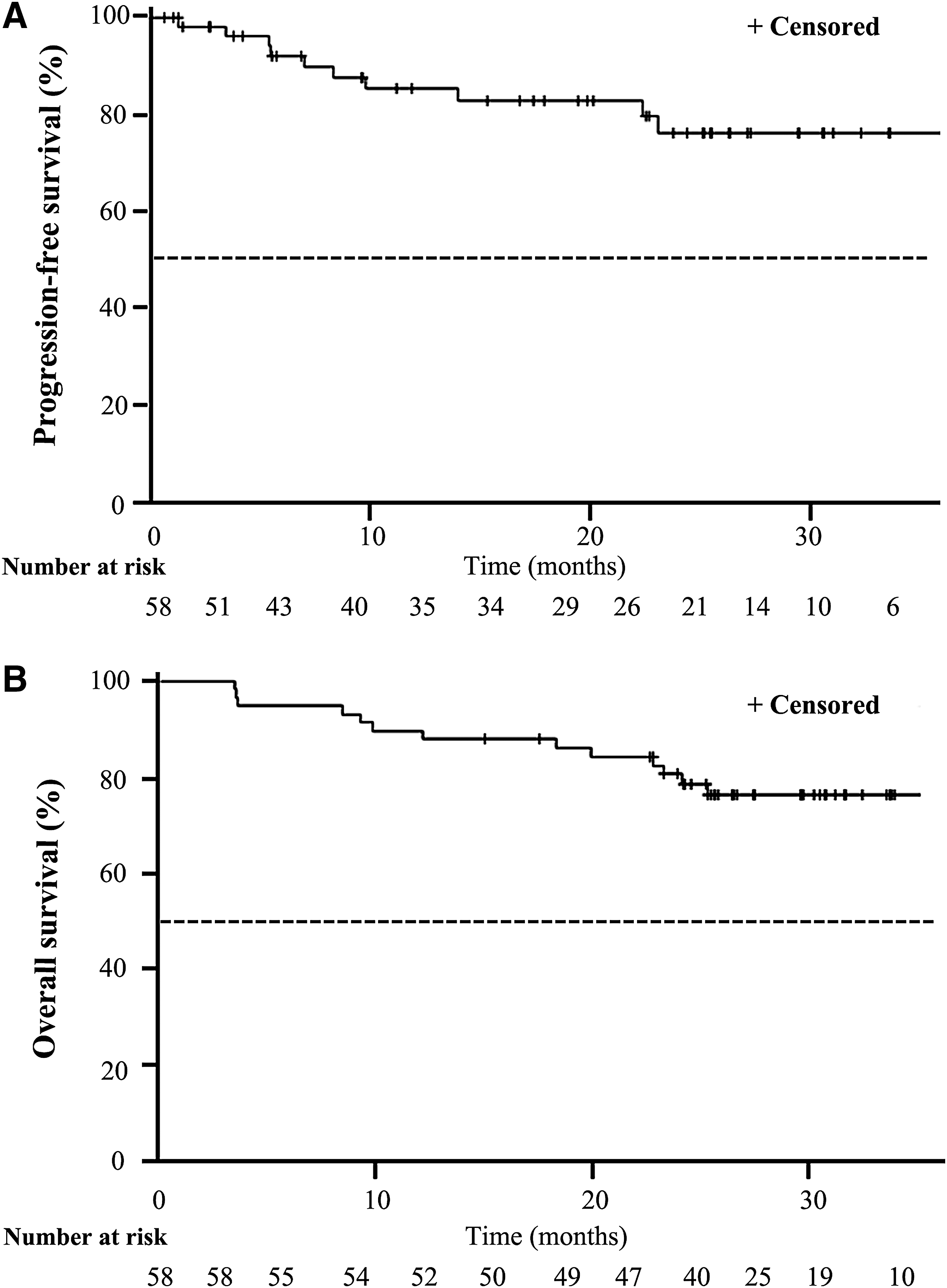
The median follow-up time was 9.8 months. In the FAS, the ORR was 56.90% (33/58 [confidence interval (CI) 44.15–69.64]; Fig. 1). The DCR was 93.1% (54/58 [CI 88.09–99.58]). Durable stable disease was observed in most patients, and the PFS rates at 24, 36, and 48 weeks were 92.2%, 87.8%, and 84.5%, respectively. The median PFS has not yet been reached at the time of analysis (Fig. 2A). The survival rates at 12, 24, and 36 months were 89.7%, 78.6% and 76.4%, respectively. The median OS has not yet been reached at the time of analysis (Fig. 2B).
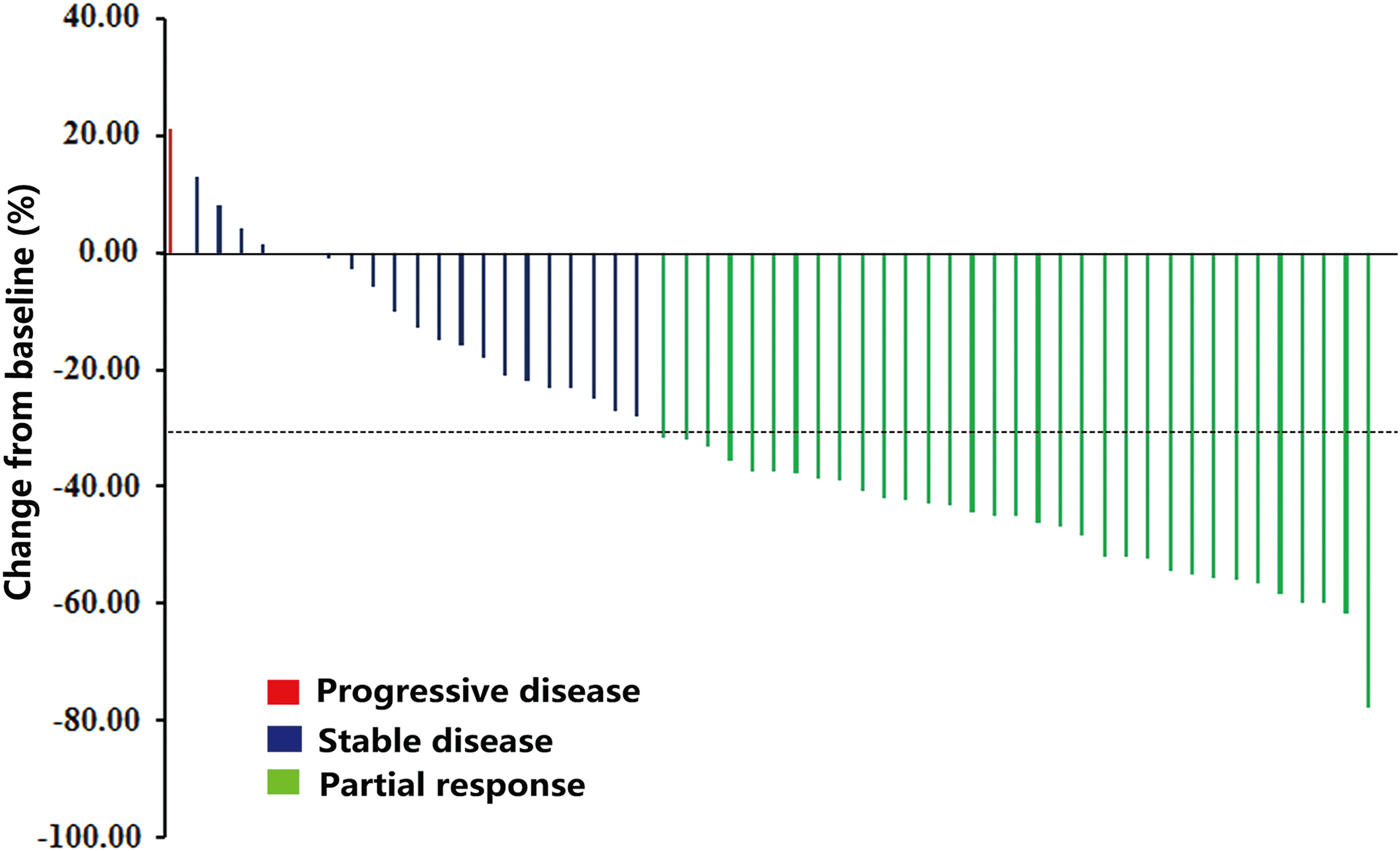
Tumor shrinkage per investigator review. Maximum reduction from baseline (or smallest increase from baseline for patients with no reductions) in the sum of the longest diameters of target lesions. The change from baseline in tumor measurement as assessed by investigator review is shown for 55 patients (per protocol set). The dotted line represents the threshold for partial response (>30% reduction from baseline sum of longest diameters). Target lesions were defined according to RECIST 1.1. Color images available online at
(
Calcitonin response
Calcitonin response at week 12 was evaluable in 51 (87.9%) patients. The most common reason that patients were not evaluable was the lack of a week 12 assessment. Significant decreases from baseline in serum calcitonin (≥50%) occurred in 45 (57.5%) patients. In the 33 patients who achieved PR, 22 had significant calcitonin decreases, ranging from 96.9% to 50.7%.
Toxicity
The median duration of anlotinib therapy was 12 months (range 0.75–22.5 months). All patients were evaluable for toxicity. Six (10%) patients discontinued treatment because of an AE. Table 2 summarizes AEs that occurred in anlotinib-treated patients (>15%). The most common AEs (all grades, ≥30%) included hand-foot syndrome (79.31%), hypertriglyceridemia (46.55%), elevated cholesterol levels (43.1%), fatigue (41.38%), proteinuria (39.66%), hypertension (39.66%), sore throat (37.93%), diarrhea (34.48%), and anorexia (34.48%). The most common grade 3 AEs (≥3%) included hand-foot syndrome (8.62%), hypertension(5.17%), hypertriglyceridemia (3.45%), elevated cholesterol levels (3.45%), and an increased direct bilirubin (3.45%). No grade 4 or grade 5 AE was recorded. Asymptomatic grade 1 or grade 2 QTc prolongation was observed in two patients, which subsequently normalized without further treatment.
TSH, thyrotropin; DBIL, direct bilirubin; ALT, alanine aminotransferase; TBIL, total bilirubin; LDL, low-density lipoprotein.
AEs were generally managed with dose interruptions and dose reductions, with 20.7% (12/58) of anlotinib-treated patients having dose reductions. Seven serious AEs were reported, but only one lacunar infarction was considered by the investigator to be probably related to anlotinib.
Discussion
Given its rarity, it is challenging to enroll MTC patients in clinical trials. Currently, only vandetanib and cabozantinib have been approved for MTC treatment by the FDA. However, due to lack of clinical evidence to support their use in Chinese patients, both vandetanib and cabozantinib have not been approved in China. Anlotinib has demonstrated promising antitumor effects in patients with MTC in a Phase I study (27). The Phase II study presented here further confirms the antitumor activity of anlotinib in patients with locally advanced or metastatic MTC. Partial remissions were observed in 56.9% of the patients, and the PFS rate at 48 weeks was 85.5%. Besides, the biochemical response rate for calcitonin was 57.7%. All these data suggest clinically meaningful tumor control. The OS data cannot be determined yet, and the final assessment will be done when 50% of patients have died.
It is notable that approximately 80% of the patients included in this anlotinib study were naïve to any systemic treatment, and they reflect a patient population with relatively indolent disease compared to those with progressive disease after one or more systematic treatment(s). This may partly explain the similar antitumor effect of anlotinib in this study with vandetanib in a Phase III study, which included a comparable patient population (3). In contrast, the Phase III study of cabozantinib enrolled patients with pretreated and progressive MTC and showed a lower ORR of 28% and a shorter median PFS of 11.2 months (21).
Treatment with anlotinib was generally well tolerated. The majority of AEs were manageable according to standard clinical practice alone or in combination with anlotinib dose interruptions or dose reductions. The high frequency of lipid metabolism dysfunction including hypertriglyceridemia and cholesterol elevations was consistent with safety data in the Phase I study of anlotinib, most of which were asymptomatic and reversible. However, careful monitoring of lipids is required.
The toxicity profile of anlotinib was different compared to that of cabozantinib and vandetanib. Diarrhea was the most common AE with these drugs and occurred in >50% of the patients treated with the two agents (3,21,23). In contrast, only 22.4% of the patients receiving anlotinib experienced diarrhea, typically of grade 1 or grade 2 severity. QTc prolongation was one of the most common grade 3 or grade 4 AEs for vandetanib, which was observed in 8% of patients in the experimental arm (3), whereas only two patients receiving anlotinib were found to have asymptomatic grade 1 or grade 2 QTc prolongations, and both subsequently normalized without further treatment.
In conclusion, anlotinib demonstrated promising efficacy as well as a manageable adverse event profile in this study, which suggests that anlotinib might provide a novel effective therapeutic option for patients with advanced or metastatic MTC.
Footnotes
Acknowledgments
We thank the patients and investigators who participated in this study. We thank Dr. C.H. Zhao and Dr. F. Du for their assistance during the submission and revision process. This study was sponsored by the Jiangsu Chia-tai Tianqing Pharmaceutical Co. Ltd. The funders had no role in the design, data collection, or analysis. The corresponding author had full access to the data and took final responsibility for the decision to submit for publication.
Author Disclosure Statement
The authors declare that they have no competing interests.