
Abstract
Background:
Constitutively active thyrotropin receptor (TSHR) mutations are the most common etiology of non-autoimmune hyperthyroidism (NAH). Thus far, the functionality of these mutations has been tested in vitro, but the in vivo models are lacking.
Methods:
To understand the pathophysiology of NAH, the patient-derived constitutively active TSHR D633H mutation was introduced into the murine Tshr by homologous recombination.
Results:
In this model, both subclinical and overt hyperthyroidism was observed, depending on the age, sex, and genotype. Homozygous mice presented hyperthyroidism at two months of age, while heterozygous animals showed only suppressed thyrotropin. Interestingly, at six months of age, thyroid hormone concentrations in all mutant mice were analogous to wild-type mice, and they showed colloid goiter with flattened thyrocytes. Strikingly, at one year of age, nearly all homozygous mice presented large papillary thyroid carcinomas. Mechanistically, this papillary thyroid carcinoma phenotype was associated with an overactive thyroid and strongly increased stainings of proliferation-, pERK-, and NKX2-1 markers, but no mutations in the “hot-spot” areas of common oncogenes (Braf, Nras, and Kras) were found.
Conclusions:
This is the first study to reveal the dynamic age-, sex-, and genotype-dependent development of NAH. Furthermore, the study shows that a constitutively active TSHR can trigger a malignant transformation of thyrocytes.
Introduction
H
The activation of the TSHR via thyrotropin (TSH), TSHR autoantibodies, or an activating TSHR mutation induces an increase of iodide uptake, TH synthesis, and release via Gs- and Gq/11-mediated pathways. While the Gs pathway is the main regulator of TH synthesis, secretion, iodide uptake, and thyrocyte proliferation (13), Gq/11 signaling has also been shown to be important for iodine organification, TH release, and goiter growth in a mouse model (14). This diversity in signaling has been suggested to play a distinct role in the pathogenesis of thyroid diseases. TSHR autoantibodies (15,16) or receptor mutations can alter G protein signaling differently, resulting in different phenotypes. All known constitutively active mutations of the TSHR activate Gs and rarely also Gq/11 signaling. However, there is no clear correlation between the severity of the phenotype and the signaling of the mutant TSHR receptors in vitro (7,17). In mice, transgenic overexpression of a constitutively active G
To understand the role of TSHR signaling in the development of hyperthyroidism and thyroid growth, a knock-in (KI) mouse model was generated harboring the patient-derived TSHR D633H mutation. This mutation is located in the TSHR “hot-spot” area in transmembrane helix 6 (Fig. 1A), and has been identified in patients with TTNs (23) and in one thyroid insular carcinoma (24). Further criteria for the selection of this particular mutation were based on previously published in vitro data, which showed a comparable cell surface expression to the wild-type TSHR and a strong basal activity for Gs and a moderate basal activity for Gq/11 signaling (25).
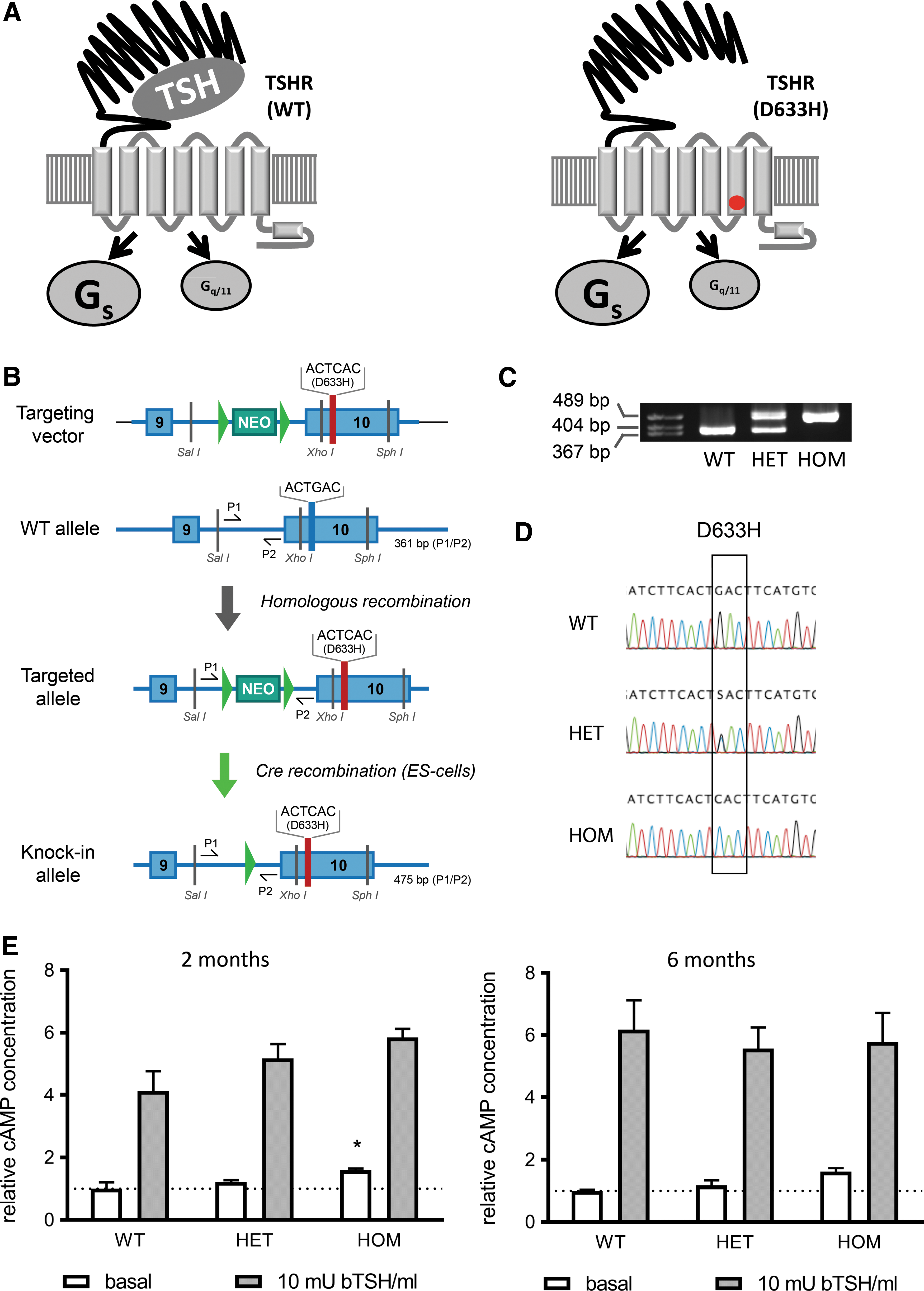
Generation of the thyrotropin receptor (TSHR) D633H mice. (
For the first time, this study reports that a constitutive TSHR activity leads to a variable development of subclinical to overt hyperthyroidism in an age-, sex-, and genotype-dependent manner in mice. Furthermore, development of PTC was observed in older TSHR D633H KI animals.
Methods
Generation of the TSHR mutants and functional characterization in vitro
The TSHR D633H variant was generated by polymerase chain reaction (PCR) mutagenesis, as previously described, using human or mouse wild-type (WT) TSHR-pSVL constructs as templates (26). The mutated TSHR sequence was verified by sequencing (ABI Advanced Biotechnologies, Inc.). Briefly, COS-7 cells grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum, 100 IU/mL penicillin, and 100 μg/mL streptomycin (Gibco Life Technologies) at 37°C in a humidified 5% CO2 incubator were transiently transfected in 12-well plates (1 × 105 cells per well) or 48-well plates (0.25 × 105 cells per well) with 1 and 0.25 μg DNA per well, respectively, using the GeneJammer® Transfection Reagent (Stratagene). The determination of TSHR cell surface expression, TSH (recombinant human TSH; Thyrogen), stimulated intracellular cAMP, and inositol phosphate (IP) levels were performed, as previously described (26,27).
Generation of TSHR D633H KI mice, animal husbandry, and genotyping
To introduce the D633H mutation into the murine Tshr locus, the nucleotide sequence GAC coding for codon 633 was replaced with the sequence CAC by homologous recombination (Fig. 1). In detail, BAC clones containing the murine Tshr gene (ENSMUSG00000020963) were obtained from BACPAC Resources Center (Children's Hospital Oakland Research Institute). An 8100 bp genomic DNA fragment spanning exons 9 and 10, intron 9, and 3′ UTR of the Tshr gene was cloned into the pACYCY177 vector (New England Biolabs) by Red/ET recombination according to the manufacturer's instructions (Gene Bridges GmbH). Site-directed mutagenesis was performed with a sense 5′-CTG TGT TGA TCT TCA CTC ACT TCA TGT GCA TGG CGC-3′ and antisense 5′-GCG CCA TGC ACA TGA AGT GAG TGA AGA TCA ACA CAG-3′ primer to generate the point mutation in exon 10 by using the QuikChange Site-Directed Mutagenesis kit (#200523; Stratagene). A Neo resistance gene was inserted and flanked by two LoxP sites with additional 37 and 25 nucleotides from the donor vector on the 5′ and 3′ side, respectively. These LoxP sites were introduced into intron 9 by Red/ET recombination. The DNA fragment containing exon 10 with the point mutation and Neo cassette was replaced with WT exon 10 in pACYC 177 backbone by Red/ET recombination. Restriction enzyme digestion and sequencing confirmed the validity of the final targeting construct. G4 embryonic stem cells (derived from mouse 129S6/C57bl/6Ncr) were cultured on neomycin-resistant primary embryonic fibroblast feeder cells. Ten million cells were electroporated with 30 μg of linearized targeting construct and cultured in the presence of 300 μg/mL G418 (Sigma–Aldrich), and 96 colonies were picked after seven to nine days of selection for further processing. For Neo cassette deletion, the targeted ES cells were electroporated with pCAGGS-Cre plasmid and cultured for three to five days, screened for the correct homologous recombination by PCR, and confirmed by sequencing. The targeted ES cells were injected into C57bl/N6 mouse blastocysts (Charles River Laboratories) to generate chimeric mice. The presence of the D633H mutation was investigated by PCR using genomic DNA with primers P1 and P2 at 63°C annealing temperature and analyzed via agarose gel electrophoresis (Fig. 1A and Supplementary Table S1; Supplementary Data are available online at
Histology, immunohistochemistry, and morphometric analysis
Formalin-fixed (10% formalin in phosphate-buffered saline [PBS]), paraffin-embedded tissue samples were cut into sections 4 μm thick and stained with hematoxylin and eosin (H&E) for histological analysis using standard methods. Stained sections were imaged with a Pannoramic Digital Slide Scanner (3D HISTECH). For the classification of thyroid neoplasia, the definition of the World Health Organization on Tumours of the Thyroid and Parathyroid was used (29). Histology was examined by two experienced pathologists (M.K. and R.G.) independently from each other.
Immunohistochemistry (IHC) was performed, as described previously (30). The following antibodies were used in the given concentrations: mouse anti-TTF-1 (anti-NKX2-1; 8G7G3/1, #M3575; Dako) 1.68 μg/mL, rat anti-mouse Ki67 0.5 μg/mL (SolA15, #14-5698-82; eBioscience), rabbit anti-mouse p44/42 MAPK (ERK1/2) 0.4 μg/mL (137F5, #4695; Cell Signaling Technology), and rabbit anti-mouse phospho-p44/42 MAPK (ERK1/2; Thr202/Tyr204) 0.25 μg/mL (D13.14.4E9, #4370; Cell Signaling Technology). Corresponding secondary horseradish peroxidase-conjugated anti-mouse and anti-rabbit antibodies and rat-on-mouse HRP polymer (#RT517; Biocare Medical) with Dako EnVision detection kits (#K500711-2; Dako) were used. From three animals per genotype and sex, the thyrocyte thickness of two neighboring thyrocyte layers was measured for 20 randomly selected follicle–follicle borders using Pannoramic Viewer v5.14.4 (3D HISTECH). In addition, this software was used to determine the intrafollicular area of 20 randomly chosen follicles; for 12-month-old animals, the follicles from PTC areas were excluded. The thyrocyte proliferation rate was determined using Fiji software (31). The number of Ki67 positive cells and total number of cells was counted in three to six not overlapping, randomly selected areas per thyroid section of three mice per age, sex, and genotype. The ratio of Ki67 positive cells to total cell number was expressed as proliferation index in percent.
Hormone measurements, cAMP determination, primary cell culture
Free thyroxine (fT4; #DNOV052) and total thyroxine (TT4; #DNOV054) serum concentrations were determined using a commercially available enzyme-linked immunosorbent assay (Novatec). Serum TSH levels were analyzed with the Mouse Pituitary Magnetic Bead Panel (#MPTMAG-49K; Merck Millipore) according to the manufacturer's instructions. Intracellular cAMP was measured using a radio-immunoassay (RIA), as previously described (32). Primary cell culture of dissected thyroids was performed according to Jeker et al. (33). For cAMP accumulation, cells were incubated at 37°C and 5% CO2 with DMEM/F12 in the presence of 0.6 mM IBMX for 2 h and with or without 10 mIU/mL bTSH (Sigma–Aldrich). Next, cells were incubated with 0.1 M HCl for 30 min on ice. HCl was evaporated, and the cAMP was re-suspended in PBS with 0.1% bovine serum albumin. For normalization, protein concentrations were determined from three wells per genotype using the Pierce BCA protein assay (#23227; Thermo Fisher Scientific).
Laser capture microdissection
Formalin-fixed (10% formalin in PBS), paraffin-embedded thyroid tissue from 12-month-old homozygous (HOM) animals (seven females) were cut into sections 4 μm thick, placed on MembraneSlides 1.0 PEN (#415190-9041-000; Carl Zeiss), and stained with H&E. An image for each thyroid section was taken at low magnification for orientation and for labeling the areas of interest. Laser capture microdissection was performed using a Zeiss laser microdissection platform PALM MicroBeam (Carl Zeiss). DNA extraction from microdissected thyroid tissue was performed with a commercial kit (#80234; AllPrep DNA/RNA FFPE kit; Qiagen) according to the manufacturer's instructions. PCR with genomic DNA was then used to amplify the following gene mutations in Braf (exon 15, codon 600/601), Kras (exon 2, codon 12/13), and Nras (exon 3, codon 61). The primers used are given in Supplementary Table S2. The purified PCR products were then sent for sequencing (GATC Biotech AG).
Quantitative reverse transcription PCR
RNA isolation from snap-frozen total thyroids and quantitative reverse transcription PCR was performed as described previously (34) for the following genes: Ctsb, Ctsl, Arrb2, Nkx2-1, Pax8, Nis, Tg, thyroid peroxidase (Tpo), and Tshr and normalized to peptidylprolyl isomerase A (Ppia) and receptor like protein 19 (Rlp19) with primers given in Supplementary Table S2.
Study approval
All experiments were authorized by the National Animal Experiment Board of Finland (License number: 10266).
Statistics
GraphPad Prism v7 (GraphPad Software, Inc.) was used for statistical analysis. An unpaired t-test and one-way analysis of variance with Dunnett's post hoc test and nonparametric Kruskal–Wallis test were used to determine statistical significances. A p-value of <0.05 was set as the limit of statistical significance.
Results
Mouse and human TSHR D633H mutations have a similar constitutive activity in vitro
The TSHR D633H mutation has been identified in several patients with hyperthyroidism (23,24) (
TSHR D633H mutation leads to an increased cAMP concentration in mouse primary thyrocytes
To study the physiological consequences of TSHR D633H mutation, a knock-in mouse line was generated harboring the TSHR D633H mutation in the corresponding mouse Tshr locus. Classical gene targeting in mouse embryonic stem cells with homologous recombination technique was used to exchange the nucleotide sequence GAG coding for aspartic acid (D) at codon 633 with the sequence CAC coding for histidine (H; Fig. 1B). Chimeric mice were obtained from two different embryonic stem-cell clones. The presence of the mutant Tshr allele was screened by PCR (Fig. 1C) and confirmed by direct sequencing of the targeted region using genomic DNA from WT, heterozygous (HET), and HOM mice (Fig. 1D). HET breeding showed a normal Mendelian distribution of the genotype (WT:HET:HOM = 1:1.8:1.2), sex (female:male = 1:0.9), and normal litter size (6.1 ± 2.4 pups/litter).
To test if the TSHR D633H mutation leads to an increased constitutive activity in vivo, cAMP levels were measured in primary thyroid cell cultures from two- and six-month-old mice (Fig. 1E). In two-month-old mice, cAMP levels were slightly but significantly upregulated in HOM mice compared to the WT thyrocytes. A similar trend of increased basal cAMP production was seen in six-month-old HOM mice versus controls. Furthermore, thyrocytes from two- and six-month-old WT, HET, and HOM mice responded similarly to TSH stimulation, indicating a normal TSHR expression and function in WT and TSHR mutants.
TSHR D633H mice develop age-, sex-, and genotype-dependent hyperthyroidism
As hyperthyroidism may lead to weight loss and accelerated growth, these parameters were monitored over 12 months. In overall phenotypic analyses, no obvious differences in body or tail lengths were noticed over the investigated period of 12 months (Table 1 and Supplementary Fig. S2A and B). However, after the first six months, HOM males and females did not gain as much weight as the WT littermates (Fig. 2A). Thus, at 12 months of age, the body weights of HOM males and females were >25% lower than in their WT littermates (Table 1 and Fig. 2A). There were no significant differences in body weights at 2 or 16 days of age between WT, HET, and HOM mice (data not shown).
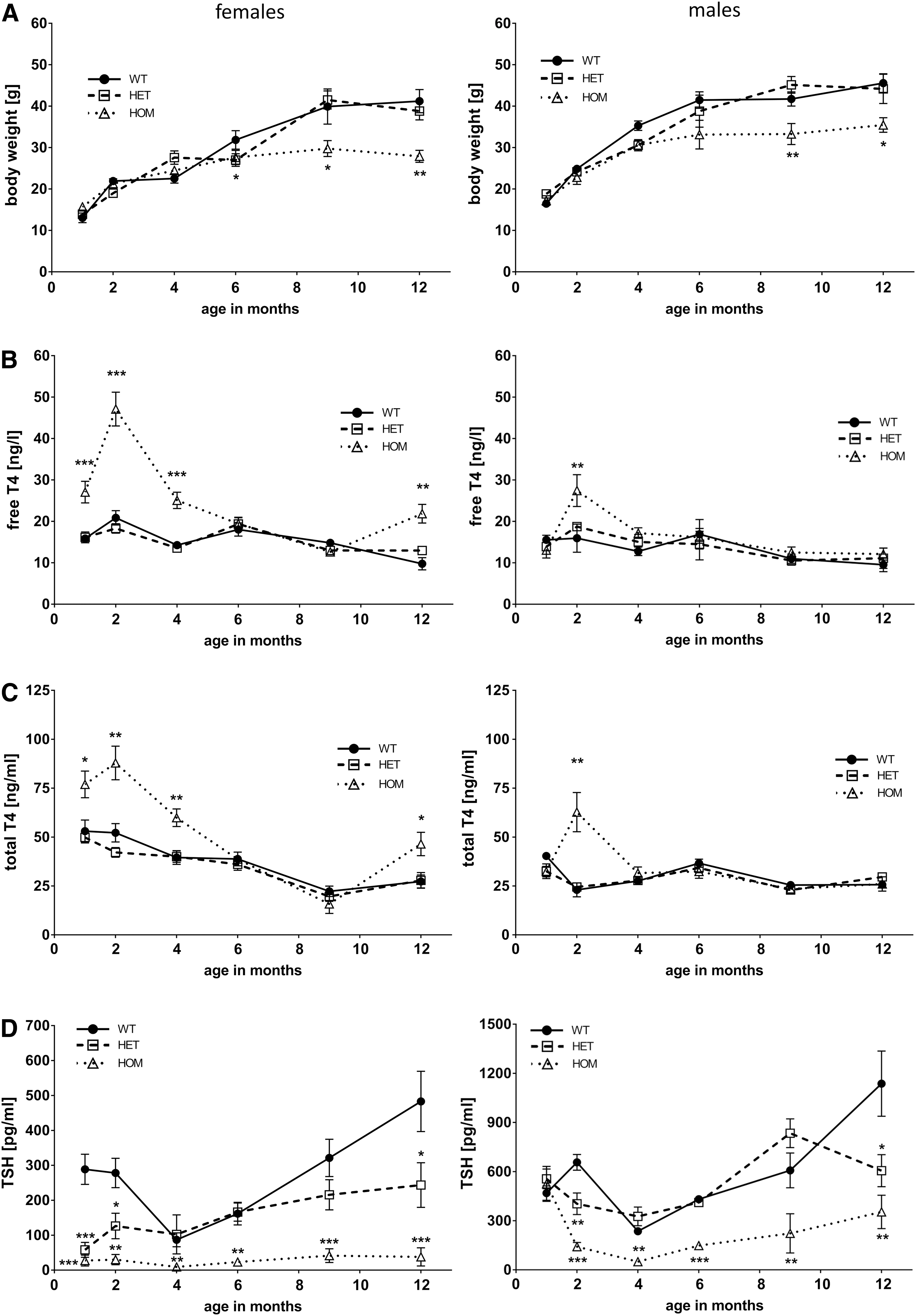
Body-weight development and thyroid function tests of WT, HET, and HOM TSHR D633H mice. (
Overview of body parameters (n = 6–7), thyroid morphology (n = 3), and litter size (n = 6) in WT mice compared to HET and HOM TSHR D633H mice. Litter size determination was not applicable (N/A) for male mice. Data presented as mean ± SEM.
p < 0.05; **p < 0.01; *** p < 0.001. Statistical analysis was carried out using the one-way ANOVA nonparametric Kruskal–Wallis module.
TSHR, thyrotropin receptor; WT, wild type; HET, heterozygous; HOM, homozygous; SEM, standard error of the mean; ANOVA, analysis of variance.
To understand the impact of the TSHR D633H on thyroid physiology, serum TSH and TH concentrations were measured at different time points during the 12-month life-span. Interestingly, a dynamic age-, sex-, and mutant allele–dependent development of hyperthyroidism was detected. Despite the embryonic onset of TSHR expression (starting embryonic day E15) (36) and the initiation of TH synthesis thereafter, no obvious signs of congenital hyperthyroidism were evident in TSHR D633H mice at any age group analyzed (Fig. 2). However, compared to the WT littermates, the HOM females had significantly elevated serum TH concentrations between one and four months of age. Serum TH levels were highest in HOM females at two months of age, with a 2.3-fold increase in fT4 and a 4.2-fold increase in TT4 (Fig. 2B and C). HOM males showed overt hyperthyroidism only at two months of age, with a 1.7- and 2.7-fold increase in serum fT4 and TT4, respectively (Fig. 2B and C). Suppressed TSH serum concentrations were measured at all time points for HOM females. Surprisingly, TSH values at one month of age were not altered for HOM males but were significantly reduced starting at two months (Fig. 2D). In general, HOM male mice had higher TSH levels than HOM females at each time point measured. HET females and males displayed decreased serum TSH but unaltered fT4 and TT4 levels (Fig. 2). Unexpectedly, no difference in serum fT4 or TT4 levels in HET or HOM females and males was observed at six months of age (Fig. 2B and C), despite the suppressed serum TSH levels in HOM animals of both sexes (Fig. 2D). Interestingly, overt hyperthyroidism was again present in one-year-old HOM female mice (Fig. 2B, C, and D). Moreover, serum TSH concentrations were constantly increasing after four months, which was most evident for WT and HET mice and less pronounced for HOM males (Fig. 2D). Together, these data show that a constitutively active TSHR leads to hyperthyroidism, but it can remain compensated in males, HET animals, and also temporarily in HOM females.
TSHR D633H mutant mice develop colloid goiter associated with thin thyroid epithelia
Based on the identification of the TSHR D633H from patients with thyroid adenomas, the thyroid growth and histology of TSHR D633H mice was analyzed. In general, thyroids were larger in HET and HOM animals of both sexes at all time points compared to their WT littermates. Already at two months of age, HOM mice presented with a significantly increased thyroid weight (2.8- and 2.3-fold increase in female and male HOM mice, respectively, compared to WT littermates; Fig. 3A). The thyroid weight increased progressively with age in TSHR D633H KI mice. At six months of age, the thyroid weight in females and males was 2.0- and 2.2-fold higher in HET animals and 3.7- and 2.5-fold higher in HOM mice compared to WT littermates (Fig. 3A). Histological examination of thyroids at two months of age revealed only marginal alterations (Fig. 3B, upper panel). The intrafollicular area was 2.8-fold larger and thyrocyte thickness was 15% smaller in HOM females compared to WTs. Similarly, the intrafollicular area in HET and HOM males was 2.0- and 2.2-fold larger than in WTs, respectively, but thyrocyte thickness was not altered (Fig. 3C and Table 1). However, at six months of age, all mice of both sexes showed follicles with very thin, flattened thyrocyte epithelium and bigger follicle sizes (Table 1). The large follicles appeared also in 12-month-old TSHR D633H animals, regardless of the genotype and sex.
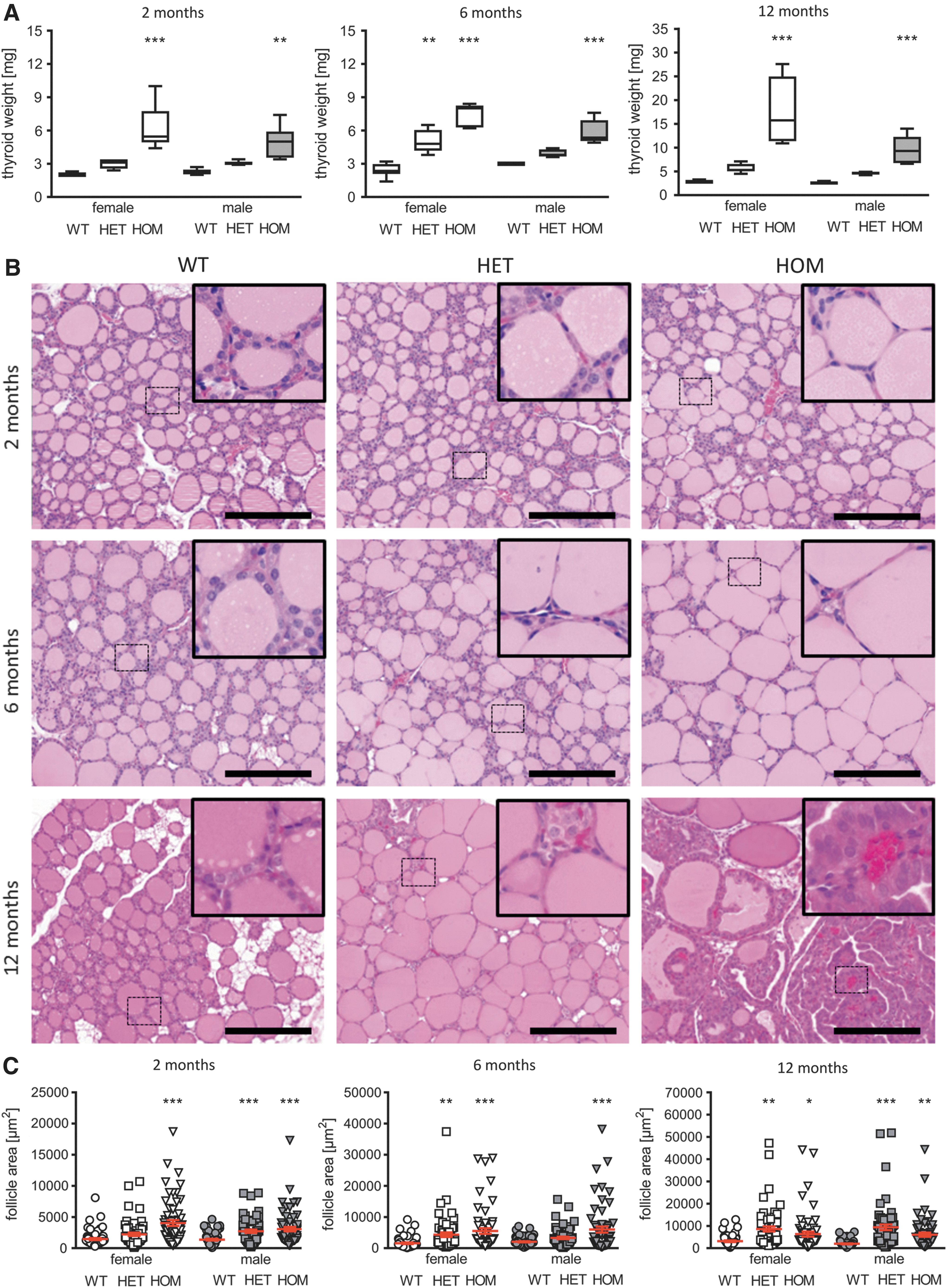
Morphologic alterations of the thyroid gland in HET and HOM TSHR D633H mice. (
Long-term activation of TSHR leads to PTC in TSHR D633H mutant mice
To determine the long-term impact of constitutive TSHR activity on thyroid growth, thyroid weight and histology were analyzed in one-year-old mice. Surprisingly, 12-month-old HET and HOM mice developed thyroid neoplasia with thick thyrocyte epithelium and protrusions of the thyrocyte layer into the lumen of the follicle (Fig. 3B, lower panel). Further histological analysis exhibited typical characteristics of PTC comprising papillae with distinct fibrovascular cores, nuclear clearance, nuclear grooves, and pseudo-inclusions as well as overlapping nuclei (Fig. 4A and B). PTCs were detected in 88% (7/8) of HOM females and in 71% (5/7) of HOM males, as well as in about 30% (2/7) of HET females and in one HET male (1/5 mice; Fig. 5A and Supplementary Fig. S3). The proportion of the PTC area in comparison to the whole thyroid section was on average 50% in HOM males and females (Fig. 5B and C), and clearly smaller in HET animals (Fig. 5B). In line with the development of PTC, the thyroid weight increased strongly after six months in HOM animals, whereas the increase in thyroid weight gain before that age was only marginal (Fig. 3A). Furthermore, PTCs of 12-month-old HOM mice presented a high proliferation rate, as shown by strong Ki67-positive staining (Fig. 5D). Quantification of Ki67-positive cells showed that 12-month-old HOM animals have a significantly increased proliferation index compared to WT thyroids (14–17% in HOM animals vs. 2% in WTs; Fig. 5E). Lower but significantly increased proliferation was also determined in HET females at 12 months of age (Fig. 5E). In general, the TSHR D633H KI mice had a higher thyrocyte proliferation rate, and the proliferation index in the mutant mice was already slightly increased at two months and six months of age (Fig. 5E). IHC for NK2 homeobox 1 (NKX2-1) showed that all PTC-forming cells express NKX2-1, which indicates maintenance of thyrocyte differentiation also in neoplasia (Fig. 5F). Furthermore, an activation of MAPK pathway in the PTCs was indicated by abundant staining of phosphorylated ERK1/2 in the majority PTC lesions of the TSHR D633H mice (Fig. 5H).
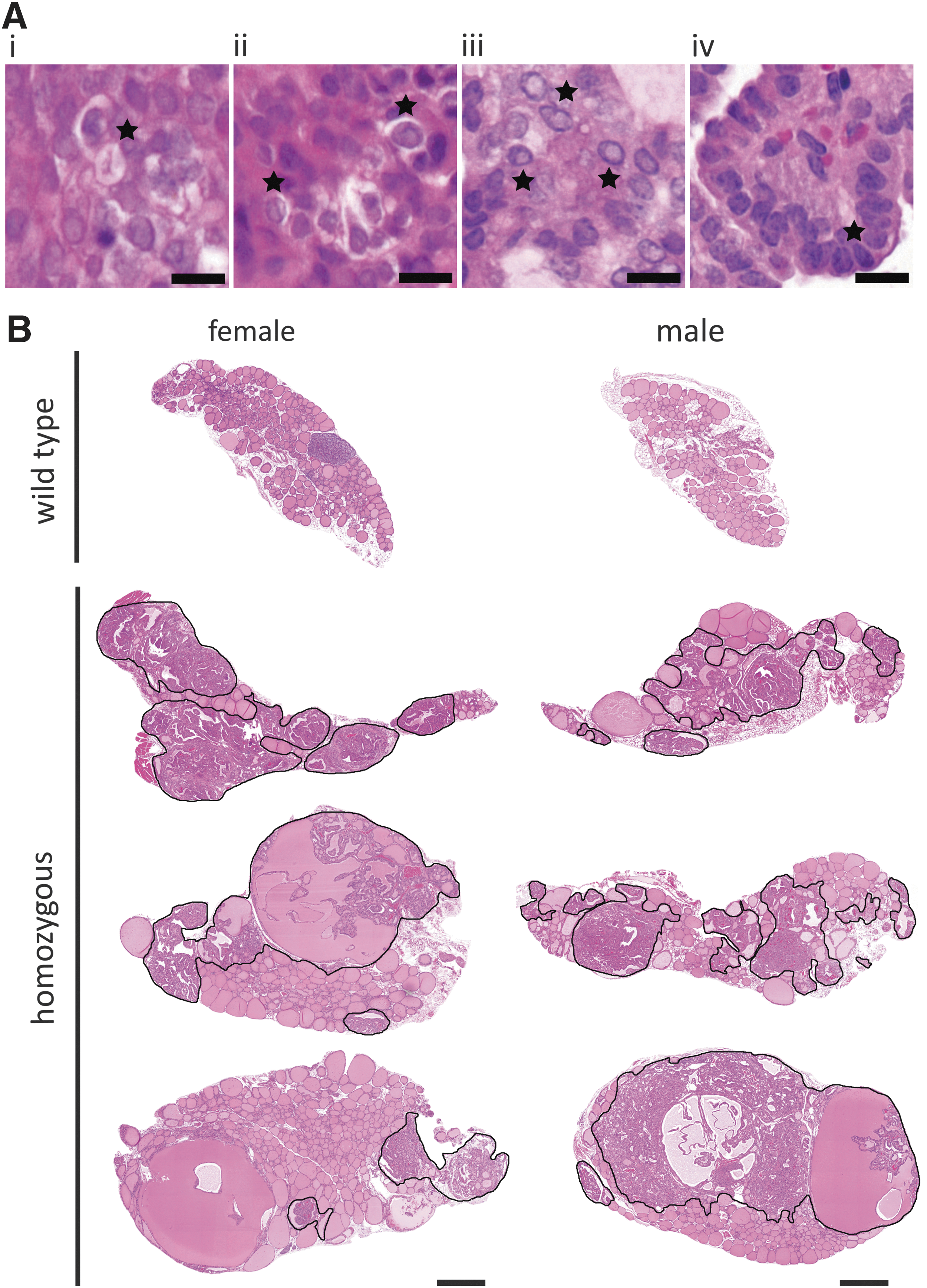
Papillary thyroid carcinoma (PTC) in TSHR D633H mice at 12 months of age. (
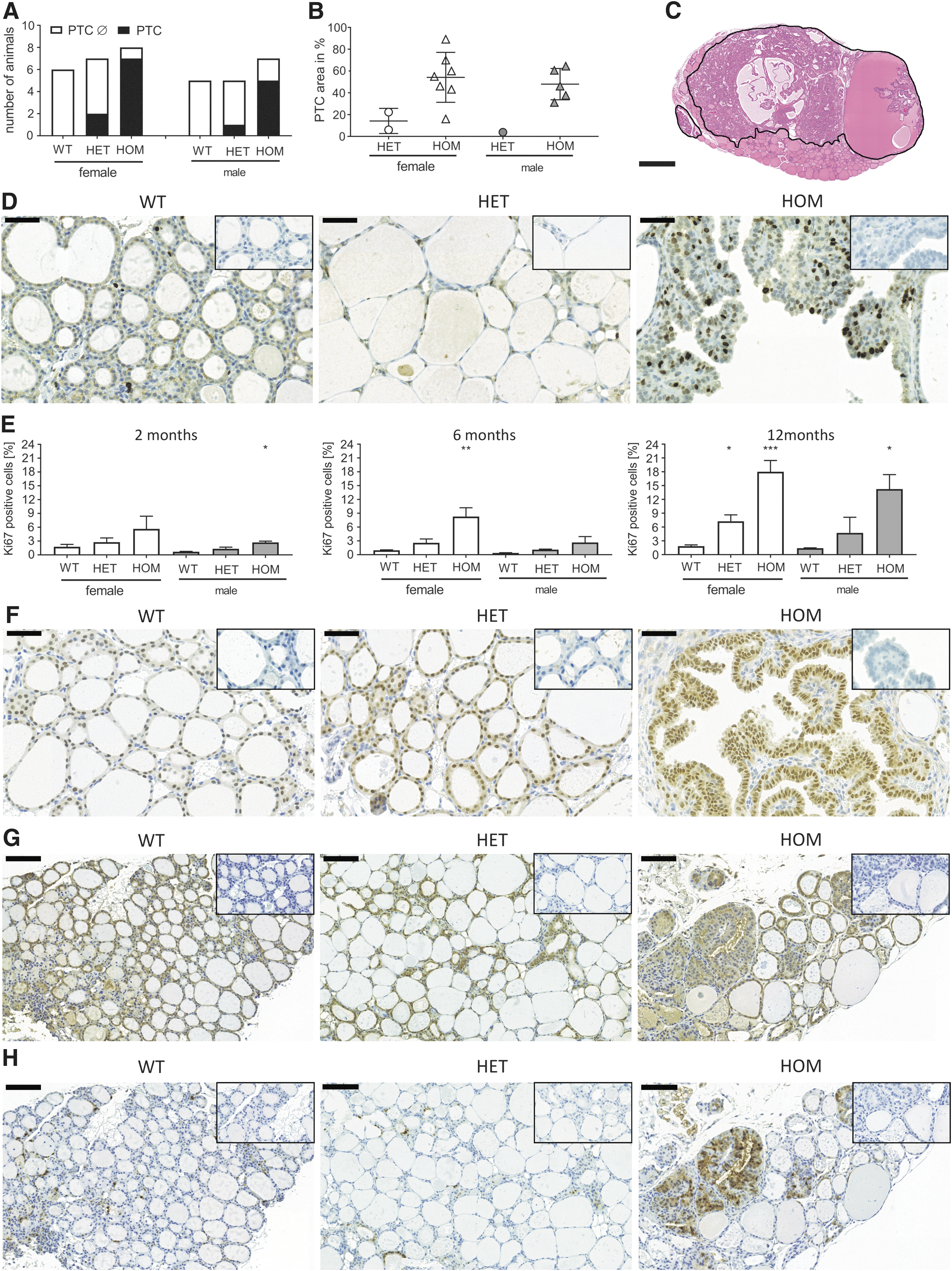
Analyses of PTC in 12-month-old TSHR D633H animals. (
In humans, mutations in BRAF, KRAS, or NRAS can be found in up to 70% of PTCs (37). To test, if the KI model harbors any mutations in the “hot-spot” areas of these genes, genomic DNA from micro-dissected PTC lesions was isolated and sequenced. However, the DNA from the PTC areas of TSHR D633H mice revealed no mutations in 19 tumors tested for Braf mutations, 13 tumors tested for Kras mutations, and 9 tumors tested for Nras mutations.
Determination of expression levels of thyroid-specific genes
To reveal the possible compensatory mechanisms in the dynamic development of hyperthyroidism in TSHR D633H KI mice, the expression of thyroid-specific genes was analyzed by quantitative PCR. Despite the constitutive activation of the mutant TSHR, the differences in serum TSH and hyperthyroidism in different sex and genotypes, no difference was detected between HET and HOM mice compared to WTs for Tshr mRNA at any time point (Supplementary Fig. S4). At two months of age, a differential expression of the analyzed thyroid-specific genes was only seen in HOM male thyroids, in which the expression of paired box 8 (Pax8) and sodium–iodide symporter (Nis) was significantly increased compared to WT controls. At six months of age, when the TH concentrations in both sexes of HOM and HET TSHR D633H KI animals were comparable to WT, no differences in the gene expression were seen, except for decreased thyroglobulin (Tg) mRNA expression in mutant females (Supplementary Fig. S3). The 12-month-old animals revealed the most prominent changes in thyroid-specific gene expression, most likely reflecting the changes in the large PTC areas seen in HET and HOM TSHR D633H mice. Additionally, a clear sex difference was observed. In 12-month-old HET and HOM female mice, the mRNA levels for Pax8 and Tg were downregulated, while in HOM females beta-arrestin 2 (Arrb2) and cathepsin B (Ctsb) were upregulated and Nis was downregulated. In contrast, HOM males showed an upregulated expression for Nkx2-1 and cathepsin L (Ctsl), whereas HET male mice did not show any changes (Supplementary Fig. S4).
Discussion
There is still a knowledge gap concerning the natural development and the long-term effects of NAH. Here, a knock-in mouse model carrying the constitutively active TSHR mutation D633H was generated. This mutation was first identified in patients with TH producing hot thyroid nodules (23,24). The results reveal that the constitutively active TSHR leads to dynamic age-, sex-, and genotype-dependent development of NAH, and triggers the development of PTC in mice. HOM TSHR D633H female mice presented with hyperthyroidism soon after birth, which then ameliorated to euthyroid TH levels at six months of age. However, concomitantly with the appearance of PTC, TH levels rose again at one year of age. The hyperthyroidism phenotype is clearly milder in HOM males than females, with only a transient form of hyperthyroidism at an early age. The variability and compensation of hyperthyroidism in the model with the same genetic background suggests different thyroidal adaption mechanisms between males and females. In contrast to most human patients with long-standing NAH due to HET TSHR activating mutations, the HET mice in the current study presented only suppressed TSH levels. In humans, subclinical hyperthyroidism has been rarely reported in family members with inherited NAH due to HET TSHR germline mutations (38,39). Moreover, two families with FNAH have been reported harboring TSHR mutations two residues upstream (F631S) or three residues downstream (C636W) of D633H, and one TSHR variant D633Y was identified in an infant with severe SCNAH (40 –42). Despite the close proximity of these mutations in the TSHR, the clinical presentation of affected patients was variable, including differences in the onset of hyperthyroidism, goiter growth, and bone age. Therefore, the manifestation of NAH due to a TSHR mutation is a complex process, and so far, no clear correlation exists between the NAH phenotype and the TSHR genotype (17,43). This prompts the question of whether there are some yet undiscovered mechanisms to prevent TSHR-mediated NAH in mice. However, it must be kept in mind that the NAH and PTC phenotypes described here could also result from the specific feature of the TSHR D633H mutation, which can constitutively activate both the Gs and Gq/11 pathways (25).
The TSHR D633H mutation selected for this mouse model was found in adult patients with hot nodules and overt hyperthyroidism (23,24) (
The normalization of TH levels in HOM TSHR D633H mice at six months of age could be caused by several counteracting mechanisms leading to a reduced cAMP response, such as downregulation, desensitization, and/or internalization of the TSHR. Specifically, G protein coupled receptor kinases (GRKs) and ARRB2 have been shown to lower the number of receptors on the cell surface and simultaneously reduce intracellular cAMP levels (45,46). Furthermore, an increased phosphodiesterase activity has been described in human hot thyroid nodules (44). Accordingly, an increased expression of Arrb2 mRNA was detected in the 6- and 12-month-old HOM females (Supplementary Fig. S3). This is in line with an increased ARRB2 expression shown in human hot thyroid nodules and might reflect a counter-regulatory mechanism leading to partial desensitization of the TSHR (47).
In general, the acute overall physiological responses to hyperthyroidism in the TSHR D633H KI mice were rather mild. Although HOM females had elevated serum TH between one and four months of age and males temporarily at two months of age, no obvious differences in weight gain or growth (body or tail length) were noticed during the first six months of age. This differs markedly from the symptoms observed in young hyperthyroid patients in which weight loss and increased growth rate are classical symptoms (2,48). However, after six months of age, HOM animals of both sexes did not gain weight as much as WT littermates. This diminished weight gain suggests delayed tissue-specific responses to TH, or other metabolic adaptation processes, which require further investigation.
As TSH is the main factor in the diagnosis and follow-up of thyroid disorders, the serum TSH concentrations were closely monitored in the hyperthyroidism model. One obvious finding was prolonged TSH suppression in HOM mice, despite the normalization of TH levels after four months in males and after six months in females. This phenomenon has also been documented in humans after recovery of hyperthyroidism in Graves' disease and NAH (49 –52). The reason for prolonged TSH suppression is most likely to be found in the complex regulation of the hypothalamus–pituitary–thyroid axis. One possible explanation might come from the functional expression of the TSHR in folliculo-stellate cells of the pituitary as part of an ultra-short feedback loop, which, upon TSH binding, suppresses its own synthesis in thyrotrophs (53,54). The consequences of an activating TSHR mutation in the pituitary may exacerbate the inhibitory effect and result in a permanent inhibition of TSH synthesis, as seen especially in HOM females. However, further experiments are necessary to assess the potential effect of an activating TSHR mutation in the pituitary and its impact on the hypothalamus–pituitary–thyroid axis.
The thyroid histology of the TSHR D633H KI mice at two months of age showed only mild morphological changes. In contrast to that, six-month-old HOM mice revealed severely altered thyroid histology with colloid goiter and a very thin and flattened thyrocyte epithelium. Thereafter, nearly all HOM and some HET of both sexes developed PTCs. In addition to the morphologic and functional remission of hyperthyroidism, the later development of PTC in TSHR D633H KI mice was surprising. The previous transgenic mouse models for hyperthyroidism led to papillary structures but did not show typical features of PTCs (18,19,55). However, the model shares some similarities with transgenic mice expressing a mutant α1B-adrenergic receptor in the thyroid (56). In these mice, the overexpression of an α1B-adrenergic receptor mutant led to the malignant transformation of thyrocytes and activation of both Gs and Gq/11 signaling pathways. Therefore, it can be speculated that the simultaneous activation of Gq/11 is an important player in tumor formation in this model.
The majority of PTCs in humans are typically characterized by a permanently active MAPK signaling cascade due to mutations in BRAF, KRAS, or NRAS (37,57). Therefore, possible mutations were screened in the “hot-spot” areas of Braf, Kras, and Nras genes in the PTC lesions, but no mutations were found. Most likely, the chronic constitutive cAMP and Ca2+ signaling via increased proliferation triggers also other growth signals and finally transformation of the thyrocytes in this model. This was supported by the expression of phosphorylated ERK1/2 characterized by IHC seen in the PTC areas of the TSHR D633H mutants, which is indicative of the activation of the MAPK pathway in these lesions. TSH signaling can converge toward a MAPK growth signal, for example via EPAC and/or RAP1B, leading to RAS activation, as shown previously in thyrocyte models (58,59). However, together with the focal and rather late appearance of the PTC, it is probable that other genetic or epigenetic events are required.
In contrast to 30% of HET TSHR D633H KI females and 20% HET males, patients with hot thyroid nodules rarely harbor thyroid cancer in the respective thyroid tissue. The published data regarding the association of thyroid cancer and hot nodules are mostly limited to case reports or series with a small number of patients. The reported probability of a hot thyroid nodule being associated with a thyroid carcinoma in or outside the hot nodule ranges between 1% and 10.3% (22). Interestingly, there are also several reports of thyroid carcinomas presenting as hot thyroid nodules (hot thyroid carcinomas). These reports identified the constitutively activating TSHR mutations M453T, I486F, L512R, A623V, F631I, T632A, T632I, D633H, and D633Y (24,60 –65). A search for RAS and BRAF mutations as well as PAX8/PPARG rearrangements has been thus far reported only for four hot thyroid carcinomas in adults out of 90 hot carcinomas reported during the last 28 years (24,61,66,67). The increased malignancy rate of hot thyroid carcinomas of children does not appear to be associated with RAS and BRAF mutations or PAX8/PPARG and RET/PTC rearrangements (68). Other genetic factors could include the recently identified mutation in one fourth of hot nodules in the enhancer of zeste homolog 1 (EZH1), which is associated with the increased proliferation of thyrocytes (69).
Taken together, the results from the TSHR D633H KI model indicate that NAH is not as stable as expected but rather a dynamic condition involving age, sex, and Tshr allele-dependent compensatory mechanisms. Furthermore, the data strongly suggest that a permanently active TSHR can lead to the transformation of thyrocytes into cancer cells.
Footnotes
Acknowledgments
This study was supported by grants by the Finnish Pediatric and Medical Foundations, EVO grant from Turku University Hospital, Academy of Finland, Sigrid Juselius Foundation (J.K.), grants from the German Research Foundation (JA1927/3-1, JA1927/3-2, JA1927/4-1; H.J.), Turku Doctoral Programme for Molecular Medicine and Biocenter Finland (H.U.), Jenny ja Antti Wihurin rahasto (H.U.), Medicinska Understödsföreningen Liv & Hälsa r.f. (C.L.), DFG (R.P. and M.E.), and Deutsche Krebshilfe (109994; M.E.) grants and the startup grant of the University of Calgary (R.P.). We thank the personnel of the Turku Center for Disease Modeling for skillful technical assistance in various stages of this study; Anna Kostiander, Taija Leinonen, and Erica Nyman for technical assistance with the histology specimens; Taina Kirjonen for hormone measurements; Ronald Ghossein for his evaluation of the PTC samples; and Andreina Kero, Alexandra Stephenson, and Samuel Refetoff for reading and editing of the manuscript.
Author Disclosure Statement
The authors declare no conflicts of interest.