
Abstract
Resistance to thyroid hormone beta (RTHβ) is a syndrome characterized by high serum levels of thyroid hormone and unsuppressed serum thyrotropin concentrations. RTHβ is caused by mutations in the thyroid hormone receptor beta (THRB) gene, which are mostly clustered in three “hot” regions along the gene. Here, a report is given on a family with RTHβ caused by a novel mutation in the THRB gene (c.1069 G>C, p.G357R) occurring outside the historically known “hot” regions.
Introduction
Resistance to thyroid hormone beta (RTHβ) is a dominantly inherited syndrome characterized by high serum levels of thyroid hormones associated with consequently unsuppressed serum thyrotropin (TSH) concentrations (1). Patients with RTHβ carry a mutation in the beta isoform of the thyroid hormone receptor gene (THRB) (1). Although signs and symptoms are variable, patients with RTHβ often have a goiter, and they can present with tachycardia, palpitations, and nervousness (2). Here, the case of a family with RTHβ carrying a novel THRB mutation is described.
Patient
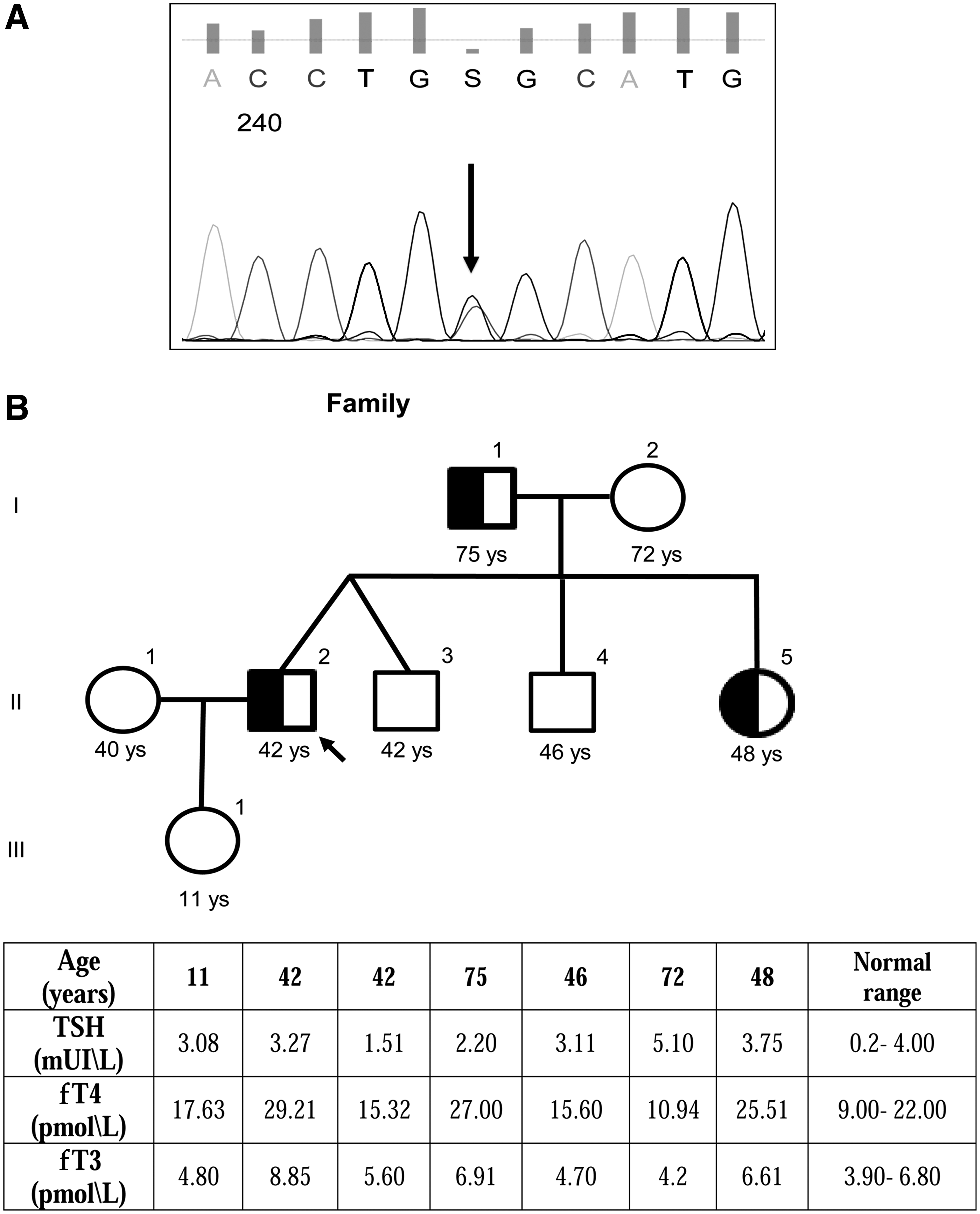
A 42-year-old Caucasian male was referred to our hospital in May 2017 with a suspected hyperthyroid state. He had a history of relapsing-remitting multiple sclerosis, anxiety and depression, and a mixed germ-cell testicular tumor (75% of embryonal carcinoma and 25% of seminoma) removed in 2010, with a negative follow-up. The patient complained of frequent palpitations and irritability. He was taking aminopyridine, gabapentin, quetiapine, alprazolam, and bisoprolol. His thyroid function test results were: free thyroxine (fT4) 29.21 pmol/L (reference range 9.00–22.00 pmol/L); free triiodothyronine (fT3) 8.85 pmol/L (reference range 3.90–6.80 pmol/L); and TSH 3.27 mIU/L (reference range 0.4–4 mIU/L; Roche, Rotkreuz, Switzerland). Antithyroglobulin and anti-TSH receptor antibodies were normal at 10 kIU/L (reference range 0–100 kIU/L; DiaSorin, Stillwater, MN) and 0.2 IU/L (reference range <1 IU/L; EuroImmun, Luebeck, Germany), respectively. Anti-thyroperoxidase levels were slightly raised (26 kIU/L; reference range 0–16 kIU/L; DiaSorin). Ultrasound showed a normal thyroid volume with increased vascularization. Magnetic resonance imaging was negative for the presence of a pituitary adenoma. The patient's sister had similar thyroid function tests and similar clinical symptoms, suggesting a diagnosis of RTH. Genetic testing was conducted to seek a germline mutation in the THRB gene. Genomic DNA was extracted from whole blood using the DNeasy Blood and Tissue kit (Qiagen, Milan, Italy) according to the manufacturer's protocol. Mutational analysis of the THRB gene (NM_ 000461.4 and P10828 exons 7–10) was performed by direct sequencing (ABI PRISM 3130; Applied Biosystems, Foster City, CA), as described elsewhere (3). The patient was found to be heterozygous for a single nucleotide substitution, a G to C transversion in exon 9, which resulted in the substitution of the normal amino acid glycine 357 with an arginine (c. 1069 G>C, p.G357R; Fig. 1). In silico analysis was performed by the following three prediction tools: Sorting Intolerant From Tolerant (SIFT;
(
The same mutation was found in the patient's father and sister, who had the same thyroid function profile. The proband's daughter had normal function test results and did not carry the mutation. Treatment was initiated with 3,3′,5-triiodothyroacetic acid (Triac) because the patient continued to complain of nervousness and palpitations, despite receiving beta-blocker treatment. This led to partial improvement of the thyrotoxic symptoms (tachycardia and nervousness) and a reduction in fT4 but not fT3 levels after two weeks of Triac therapy. These results were confirmed with up to five months of biochemical follow-up. All patients involved in this report gave their written informed consent, and the study was approved by the local ethics committee.
Discussion
This report describes a family with RTHβ caused by a new mutation occurring in an atypical location within the THRB gene. The dominant negative effect of THRB mutations responsible for this syndrome has been linked to a reduced ability to bind the ligand, T3, associated with a conserved capacity for homo- and hetero-dimerization, even with the wild-type thyroid receptor (TR) and to an aberrant ability to dissociate from co-repressor in the presence of T3 (4). The series of hydrophobic heptad repeats (“cold” region) crucial to the formation of TR homodimers and heterodimers (including co-activators and co-repressors) do not usually carry mutations that cause RTHβ. Historically, pathological mutations of THRB have been predominantly found to be clustered in three “hot” spots in the C-terminal of THRB, which is involved in T3 binding, that is, between 234 and 282 aa, between 310 and 353 aa, and between 429 and 461 aa (2). However, several mutations causing RTHβ have been reported outside these clusters between codons 353 and 374 and the so-called cold region may not be that cold after all (5). The present mutation extends cluster 2 of THRB mutations further to codon 357. This mutation probably interferes with T3 binding, as suggested by other mutations in helix 8.
Conclusions
A novel THRB gene mutation is described occurring at an atypical location in a family with RTHβ. The patient showed a clinical response to Triac treatment, although there was no evidence of any normalization of his fT3 levels, possibly due to Triac cross-reactivity in fT3 assays (6).
Footnotes
Author Disclosure Statement
The authors have no competing financial interests.