
Abstract
Background:
Anaplastic thyroid carcinoma (ATC) is one of the most aggressive and refractory cancers, and a therapy with a new concept needs to be developed. Recently, research on cancer stem cells (CSCs) has progressed, and CSCs have been suggested to be responsible for metastasis, recurrence, and therapy resistance. In ATC-CSCs, aldehyde dehydrogenase (ALDH) activity is the most reliable marker to enrich CSCs. However, it is just a marker and is not involved in CSC properties. The present study therefore aimed to identify key signaling pathways specific for ATC-CSCs.
Methods:
A small interfering RNA library targeting 719 kinases was used in a sphere formation assay and cell survival assay using ATC cell lines to select target molecules specific for CSC properties. The functions of the selected candidates were confirmed by sphere formation, cell survival, soft agar, and nude mice xenograft assays using small compound inhibitors.
Results:
The study focused on PDGFR, JAK, and PIM, whose small interfering RNAs had a higher inhibitory effect on sphere formation, as well as a lower or no effect on regular cell growth in both FRO and KTC3 cells. Next, inhibitors of PDGFR, JAK, STAT3, PIM and NF-κB were used, and all of them successfully suppressed sphere formation in a dose-dependent manner but not regular cell growth, confirming the screening results. Inhibition of the JAK/STAT3 and NF-κB pathways also reduced anchorage-independent growth in soft agar and tumor growth in nude mice.
Conclusions:
These results suggest that JAK/STAT3 and NF-κB signals play important roles in ATC-CSCs. Targeting these signaling pathways may be a promising approach to treat ATC.
Introduction
Thyroid carcinoma is the most common endocrine malignancy, and its incidence is growing worldwide. More than 90% of thyroid carcinomas are differentiated types, consisting of papillary thyroid carcinoma (PTC) and follicular thyroid carcinoma (FTC), and their overall prognosis is favorable. However, anaplastic thyroid carcinoma (ATC), which is an undifferentiated type accounting for 1–2% of all thyroid cancer cases, is one of the deadliest human neoplasms, and its mean survival is less than one year, even with multimodal treatments (1,2). To overcome this situation, a therapy with a new concept needs to be developed.
In recent years, the cancer stem cell (CSC) theory has emerged as an attractive model to explain many aspects of carcinogenesis, including metastasis, recurrence, and therapy resistance (3,4). CSCs are a small subpopulation in the cancer tissue and either self-renew or give rise to non-CSCs to produce heterogeneous tumors. Conventional chemo- and radiotherapy have been developed to target non-CSCs, but CSCs are highly resistant to these treatments. Thus, targeting CSCs is a reasonable approach to treat refractory cancers such as ATC.
In ATC, previous studies have identified several biomarkers to enrich CSCs. Among these markers, aldehyde dehydrogenase (ALDH) activity is the most reliable and widely used (5,6). Todaro et al. identified CSCs as a small subpopulation with high ALDH activity; the CSCs were highly tumorigenic in immunocompromised mice, while non-CSCs were not (5). However, the ALDH activity itself is just a marker and does not have a functional role in CSC properties (7). To target CSCs, it is necessary to identify functional molecules that are important for survival and self-renewal of CSCs, rather than a marker. The present study focused on kinases as targets because they are an important component of cell signaling pathways and can be blocked by small compounds. If inhibiting different molecules on a same signaling pathway is effective to suppress CSC properties, it indicates that the pathway is important, which increases the possibility of developing clinical applications.
This study used a small interfering RNA (siRNA) library targeting 719 kinases to screen for important molecules for CSC properties. As a result, the JAK–STAT3–NF-κB signaling cascade emerged. Several inhibitors of this pathway successfully suppressed some CSCs abilities but not growth of regular cancer cells, suggesting that this pathway is important for CSC functions and may be an attractive target to treat ATC.
Methods
Cell cultures
FRO, KTC3, and THJ16T were established from human ATCs. FRO was obtained from Dr. James Fagin (Memorial Sloan-Kettering Cancer Center, New York, NY). KTC3 was kindly provided by Dr. Junichi Kurebayashi (Kawasaki Medical School, Okayama, Japan) (8). THJ16T was obtained from Dr. John Copland (Mayo Clinic, Jacksonville, FL). ACT1 was obtained from Dr. Naoyoshi Onoda (Osaka City University, Osaka, Japan; originally established by Dr. Seiji Ohata of Tokushima University) (9). 8505C was provided by the RIKEN BRC through the National Bio-Resource Project of the MEXT, Japan. All cells were cultured in a growth medium (GM) consisting of RPMI 1640, 10% fetal bovine serum, and penicillin/streptomycin at 37°C in a humidified atmosphere with 5% CO2. Cell growth was measured using a Cell Counting Kit-8 (Dojindo). The following inhibitors were used: Imatinib (Novartis), JAK Inhibitor I (Calbiochem), STA-21 (Santa Cruz Biotechnology), AZD1208 (Selleckchem), and dehydroxymethylepoxyquinomicin (DHMEQ; synthesized by KU).
Sphere formation assay
The cells were incubated in serum-free Dulbecco's modified Eagle's medium/F-12 (1:1) supplemented with 20 ng/mL epidermal growth factor, 20 ng/ml bovine fibroblast growth factor, and B27 without vitamin A (Thermo Fisher Scientific) in a HydroCell plate (CellSeed). Spheres with a diameter of ≥100 μm were counted. Images were captured using a phase contrast microscope (Olympus). Combination drug effects on sphere formation were evaluated using CompuSyn software (ComboSyn).
siRNA screening
A MISSION siRNA Human Kinase Panel (Sigma–Aldrich) was used. This panel includes siRNAs for 719 human kinase genes. For sphere formation, cells were seeded on a 96-well HydroCell plate, and each siRNA was transfected at 10 nM using X-treme GENE siRNA transfection reagent (Roche). For each gene, three different siRNAs were mixed and used in the same well. After incubating for 96 hours, the cells were stained with Hoechst 33342 (Sigma–Aldrich), and ≥100 μm spheres were counted using an ArrayScan VTI (Thermo Fisher Scientific). For regular cell growth, cells were seeded on a regular 96-well plate, and transfection was performed as described above. After incubation for 96 hours, cell viability was determined using a Cell Counting Kit-8. For control, cells were transfected with Cy3-labeled scrambled RNA. Transfection efficiency was determined by a fluorescent microscope, and it was almost 100%.
Soft agar colony formation assay
Cells were mixed with 0.33% agar/GM and plated on a solidified 0.5% agar/GM. The agar layers were further overlaid with the GM containing appropriate concentrations of the inhibitors that were replaced every two to three days. After incubation for 10 (FRO cells) or 20 (THJ16T cells) days, images were captured using a digital camera, and the number of colonies was counted using Fiji software (10).
In vivo xenograft experiments
All procedures were conducted in accordance with the principles and procedures outlined in the Guide for the Care and Use of Laboratory Animals of Nagasaki University with approval of the Institutional Animal Care and Use Committee. FRO cells (1 × 106) re-suspended in the GM were injected subcutaneously into both flanks of six-week-old male BALB/c nu/nu mice (CLEA Japan). Then they were randomly assigned into three groups. Tumor volumes were calculated according to the formula: a 2 × b × 0.4, where a is the smallest tumor diameter and b is the diameter perpendicular to a. STA-21 or DHMEQ solution in dimethyl sulfoxide/phosphate-buffered saline (ratio 1:1) was injected intraperitoneally daily for one week, beginning from day 1 after tumor cell implantation. Control group mice received vehicle injections only.
ALDEFLUOR assay
To measure the ALDH activity, cells were labeled using an ALDEFLUOR assay kit (Stemcell Technologies, Inc.) following the manufacturer's protocol. The cells were then analyzed using a FACSJazz cell sorter (BD Biosciences). The data were further processed with FlowJo software.
Statistical analysis
Differences between groups were examined for statistical significance with one-way analysis of variance followed by Tukey's post hoc test. A p-value ≤0.05 was considered statistically significant. Data were analyzed with PRISM v6 (GraphPad Software).
Results
siRNA screening to identify important cell signaling for CSC properties
It has been demonstrated that sphere formation assay is valuable to evaluate CSC properties (3,4,6). In a previous report using eight thyroid cancer cell lines, the ability of sphere formation perfectly corresponded to that of tumor formation in mice, which is important evidence for the presence of CSCs (6). It is also applicable to high-throughput screening. The present study combined the use of a siRNA library for 719 kinase genes with the sphere formation assay. First, FRO cells were transfected with all siRNAs included in the library, and the sphere formation assay was performed. Cell survival was also measured in the regular growth condition after transfection. To identify specific cell signaling for CSC properties, sphere/survival ratios were calculated. When the ratio is small, it suggests that only sphere formation but not regular cell growth is blocked. The top 100 genes were selected and were further subjected to a second screening using another ATC cell line, KTC3. In the second screening, the sphere/survival ratios were equally analyzed. The siRNA target genes with the lowest ratios are listed in Table 1 (1st screening in FRO cells) and Table 2 (2nd screening in KTC3 cells). Among these genes, the focus was on PDGFR, JAK, and PIM because they are members of the cell signaling cascade depicted in Figure 1.
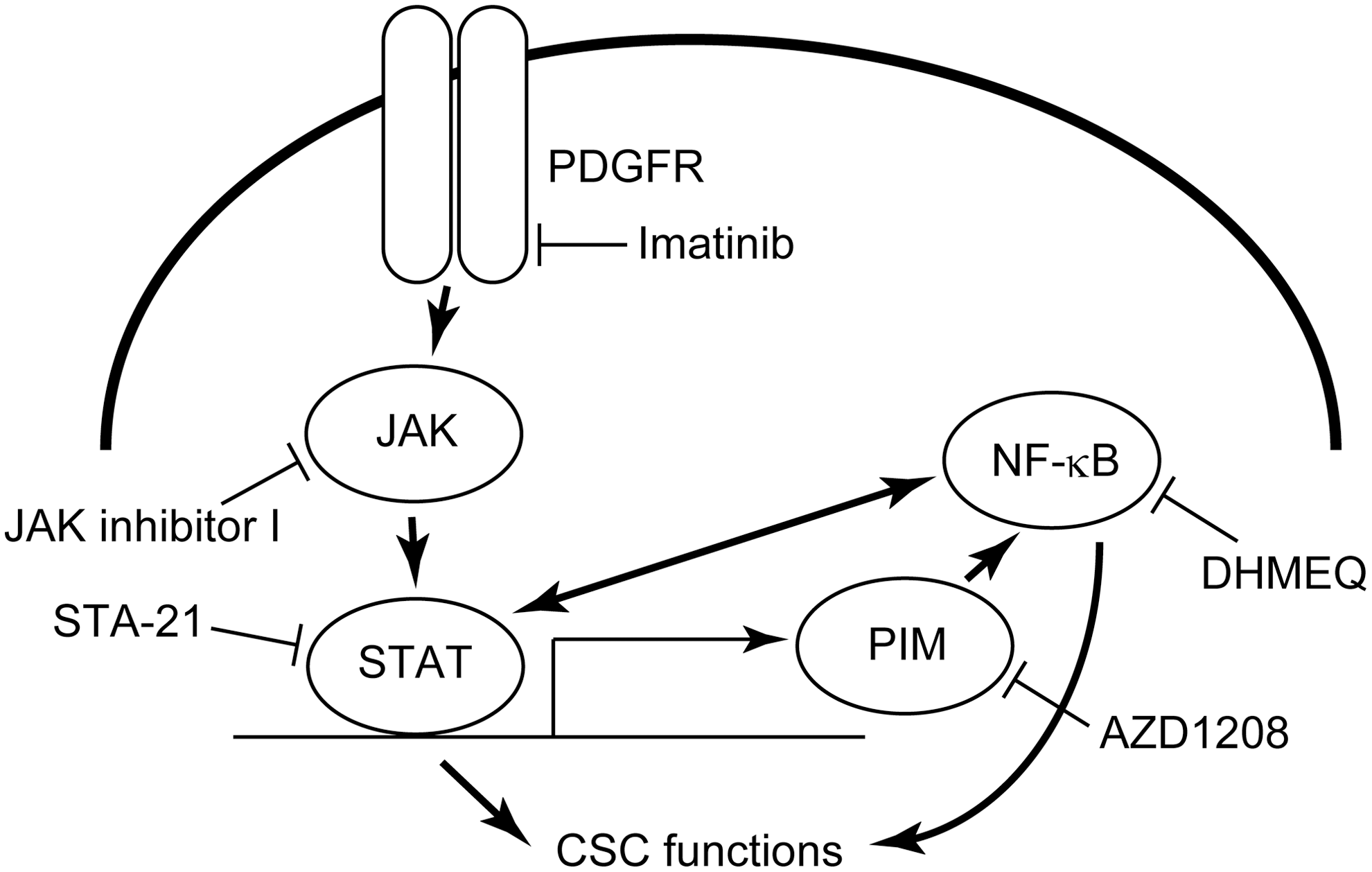
The cell signaling cascade focused on in the present study. Note the cross-talks between the STAT3 and NF-kB signaling pathways. The inhibitors used in the present study are also shown.
First Screening Using the Small Interfering RNA Library for Kinases in FRO Cells
Genes in bold were the focus of this study.
Second Screening Using the Small Interfering RNA Library for Kinases in KTC3 Cells
Genes in bold were the focus of this study.
Specific inhibitors suppressed sphere formation but not cell survival
To confirm the significance of the above signaling pathway, the cells were treated with various inhibitors, and sphere formation ability and regular cell survival were examined. The following inhibitors were used: imatinib, a PDGFR inhibitor; JAK inhibitor I, a pan-JAK inhibitor; STA-21, a STAT3 inhibitor; AZD1208, a pan-PIM inhibitor; and DHMEQ, a NF-κB inhibitor (Fig. 1). In FRO cells, all of the inhibitors suppressed sphere formation in a dose-dependent manner (Fig. 2A, left). At higher concentrations, the differences were statistically significant. On the other hand, regular cell growth was not affected at the same concentrations used in the sphere formation assay (Fig. 2A, right). KTC3 cells were also used, and similar data were obtained (Fig. 2B). Representative sphere images are shown in Supplementary Figure S1A and B. These data suggest that the signaling cascade, PDGFR–JAK–STAT3–PIM–NF-κB, has a significant role in CSC properties but not in regular cell growth.

The various inhibitors suppressed sphere formation but not regular cell growth in thyroid cancer cell lines. (
Since the JAK/STAT3 and NF-κB pathways are basically two different signaling pathways, the effect of the combination of two inhibitors, STA-21 and DHMEQ, were tested. Based on the results presented in Figure 3A, the combination index (CI) was calculated. The combination effects were synergistic in FRO cells (CI range 0.53–0.89) and almost additive in KTC3 cells (CI range 1.00–1.11).

(
The study also checked whether these inhibitors suppress sphere formation in other ATC cell lines. Although effect sizes were different, both inhibitors significantly reduced the number of spheres in 8505C and ACT1 cells (Fig. 3B).
Colony formation in soft agar
Next, to investigate the significance of the signaling pathway on the ability of anchorage-independent growth, which is also an important characteristic of tumorigenicity of cells, colony formation assays were performed in soft agar. Two inhibitors were used, JAK inhibitor I and DHMEQ, to block JAK and NF-κB, respectively. In FRO cells, both JAK inhibitor I and DHMEQ reduced the number of colonies in a dose-dependent manner (Fig. 4A, left). At higher concentrations, the differences were statistically significant. Unfortunately, colony formation in KTC3 cells was defective, even in the absence of the inhibitors. Therefore, THJ16T cells, another tumorigenic ATC cell line, were used in which sphere formation was also suppressed with the inhibitors (Fig. 4A, right). Similarly, the colony formation after treatment with JAK inhibitor I or DHMEQ was suppressed (Fig. 4A, middle). Within the range of concentrations used, the suppressive effect of JAK inhibitor I was stronger than that of DHMEQ in both cell lines (Fig. 4).

(
Tumor formation in nude mice
To examine the effect of inhibitors for the STAT3 and NF-κB pathways on in vivo tumor growth, xenograft experiments were performed using nude mice. To see the effect on tumor initiation, treatment was started from day 1 after cell implantation. Starting from day 21, the tumor size in mice treated with STA-21 or DHMEQ was significantly smaller than that in control mice (Fig. 4B).
Suppression of the JAK pathway did not alter ALDH activity
Previous studies have demonstrated that ALDH activity is the most reliable marker for CSCs in ATC cells (5,6). The ALDEFLUOR assay is a standard procedure to measure ALDH activity in each living cell. FRO and THJ16T cells were treated with JAK inhibitor I for one week, and then the ALDEFLUOR assay was performed to investigate the impact of inhibiting the pathway on the activity. Diethylaminobenzaldehyde, a specific inhibitor of ALDH, was used to measure background fluorescence. As shown in Figure 5, the treatment with JAK inhibitor I did not reduce the proportion of the ALDH-positive population in either FRO or THJ16T cells. These results suggest that ALDH function is not directly associated with CSC properties.

The JAK inhibitor I did not alter the ALDH activity. FRO and THJ16T cells were cultured in the presence of 0.5 μM of JAK inhibitor I for one week and subjected to the ALDEFLUOR assay. The cells incubated in the presence of DEAB was first analyzed as a negative control (left) to set a region to distinguish ALDH negative/positive cells, and then the test samples were measured (right). Similar results were obtained in at least two independent experiments.
Discussion
The present study successfully identified that the PDGFR–JAK–STAT3–PIM–NF-κB signaling cascade plays an important role in CSC functions in ATC. There are a number of studies reporting that STAT3 signaling is important for CSCs in a variety of cancer types such as breast cancer (11 –14), hepatocellular carcinoma (15,16), prostate cancer (17,18), lung cancer (19), ovarian cancer (20), and glioblastoma (21). However, in ATC, there is only one report showing that STAT3 plays a key role in mediating CSC properties in ATC cells (22). In this study, CSCs were enriched in the CD133-positive population, and Cucurbitacin I, a STAT3 inhibitor, suppressed the sphere-forming ability and increased sensitivities to radiochemotherapy. However, these effects were also observed in the CD133-negative cells. One possible explanation is that CD133 may not be a precise marker to select CSCs in ATC. Unfortunately, the positive rates of CD133 were not shown in this study. In a previous study, no CD133-positive cells were found in five ATC cell lines—FRO, KTC2, KTC3, ACT1, and 8505C—and it was concluded that CD133 is not a suitable maker for CSCs in ATC (6). This discrepancy cannot be explained. Since little is known about the specific cell signaling/marker in ATC-CSCs, further studies are definitely needed in this field.
Couto et al. reported that STAT3 is a negative regulator of tumor growth in PTC (23). Although they did not focus on CSCs, tumorigenesis in mice was enhanced after STAT3 inhibition, implying that STAT3 is also a tumor suppressor in PTC-CSCs. STAT3 function may be different between ATC and PTC. These findings suggest that it remains to be studied whether targeting STAT3 is effective to block anaplastic transformation from PTC to ATC.
It has been demonstrated that STAT3 inhibition reduces resistance to chemotherapy in ATC cells (24). However, this study did not separate and use the CSC fraction. In the present study, regular cell growth was not affected, but sphere formation and anchorage-independent growth were suppressed by the JAK/STAT3 inhibitors at the used concentrations. Generally, the ability of sphere formation and anchorage-independent growth reflects CSC properties, and therefore it is concluded that STAT3 signaling is important for CSC properties in ATC.
NF-κB signaling plays an important role in CSCs of various types of malignancies, including leukemia, glioblastoma, prostate cancer, ovarian cancer, breast cancer, pancreatic cancer, and colon cancer (25). However, to the best of the authors' knowledge, this study is the first to show its importance in ATC-CSCs. Indeed, NF-κB signaling is activated not only in CSCs, but also in all ATC cells (26,27). However, the present study indicates that NF-κB is crucial especially in CSCs. According to the results, there is a possibility that the treatment with low concentration of NF-κB inhibitors is effective to suppress CSC functions. Since chemoresistance of CSCs is usually high, a combination of the NF-κB inhibition and chemotherapeutics may be an attractive strategy.
There are multiple cross-talks between the JAK/STAT3 and NF-κB signaling pathways. As mentioned, the STAT3 signal is transduced to NF-κB via PIM (28,29). In addition, the activated NF-κB signal leads to production and secretion of interleukin (IL)-6, and then IL-6 activates the STAT3 signaling pathway in an autocrine/paracrine fashion (30,31). Moreover, STAT3 interacts with RelA, a p65 subunit of NF-κB, and recruits p300. Then, p300 acetylates RelA, leading to nuclear retention of RelA and thereby sustaining its transcriptional activity (32). There is a possibility that these cross-talks also influence each other in ATC-CSCs. In the present study, treatment with AZD1208 alone suppressed sphere formation substantially. However, this does not necessarily mean that the STAT3–PIM–NF-κB cascade is the most important for CSC properties in ATC because PIM also has other functions such as activating MYC and inhibiting reactive oxygen species (28). The experiments demonstrate that the effect of the combination of STAT3 and NF-κB inhibitors was synergistic in FRO cells and almost additive in KTC3 cells, suggesting that the degree of the interaction between the two pathways, JAK/STAT3 and NF-κB, depends on the cell type.
In nude mice xenograft experiments, treatment was started one day after tumor cell implantation to see the effect of the drugs on tumor initiation. Tumors treated with STA-21 or DHMEQ were statistically smaller than those in control mice after day 21, suggesting that these drugs successfully reduced the number of CSCs. Note that non-CSCs have plasticity, allowing CSCs to be generated in thyroid cancer cell lines (6,33), which may in part be involved in tumor formation in mice treated with the drugs. Nevertheless, these results support the potential clinical benefit of targeting the JAK/STAT3 and NF-κB pathways in ATCs.
As previously reported, the ALDH function itself is not involved in CSC properties in ATC (7). The present results indicate that ALDH activity is not regulated by the JAK/STAT3 signaling pathway, consistent with the above study. There may be a common upstream molecule, but further studies are still necessary to clarify the regulation of the ALDH activity in ATC-CSCs.
In conclusion, the present study demonstrates that the JAK/STAT3 and NF-κB signaling pathways play important roles in ATC-CSCs. Interference with these pathways may provide a novel approach for ATC treatment.
Footnotes
Acknowledgments
This work was supported in part by JSPS KAKENHI Grant Numbers 25861110 (M.M.) and 26293222 (S.Y.). We thank Dr. James Fagin (Memorial Sloan-Kettering Cancer Center), Dr. Junichi Kurebayashi (Kawasaki Medical School), Dr. John Copland (Mayo Clinic), and Dr. Naoyoshi Onoda (Osaka City University) for providing the FRO, KTC3, THJ16T, and ACT1 cells, respectively.
Author Disclosure Statement
The authors have nothing to disclose.
Supplementary Material
Supplementary Figure S1