
Abstract
Background:
High-mobility group box 1 (HMGB1) has been implicated in the pathogenesis of inflammatory autoimmune diseases. This study investigated the influence and mechanisms of HMGB1 in Graves' orbitopathy (GO).
Methods:
HMGB1 and its receptors (receptor for advanced glycation end products [RAGE], Toll-like receptor [TLR] 2, and TLR4) mRNA levels were evaluated by real-time polymerase chain reaction (RT-PCR) in GO and non-GO orbital tissues. The mRNA expressions of HMGB1 and its receptors were evaluated in primary cultured orbital fibroblasts from six GO patients and five healthy control subjects under interleukin (IL)-1β or tumor necrosis factor (TNF)-α stimulation using RT-PCR. HMGB1 secretions under IL-1β or TNF-α stimulation were evaluated by enzyme-linked immunosorbent assay (ELISA). The effects of an anti-HMGB1 antibody, RAGE antagonist (FPS-ZM1), and anti-TLR2 antibody on the expressions of IL-1β or TNF-α induced pro-inflammatory cytokines and phosphorylated nuclear factor kappa-light-chain-enhancer of activated B cells were evaluated using ELISA and Western blot analysis, respectively. The plasma levels of HMGB1 were compared among patients with active GO (n = 51), inactive GO (n = 48), Graves' disease without GO (n = 30), and healthy control subjects (n = 46) by ELISA.
Results:
The genes encoding HMGB1 and its receptors, as well as HMGB1 protein expression, were increased in GO orbital tissues compared to non-GO tissues. IL-1β and TNF-α stimulation increased the mRNA levels of HMGB1, RAGE, and TLR2 and the secretion of HMGB1 protein further in GO cells. Anti-HMGB1 antibody, FPS-ZM1, and anti-TLR2 antibody reduced IL-1β- or TNF-α-induced production of pro-inflammatory cytokines and phosphorylated nuclear factor kappa-light-chain-enhancer of activated B cells. The plasma levels of HMGB1 were highly increased in patients with active GO, and were significantly correlated with the clinical activity score (r = 0.566, p = 0.002) and levels of thyrotropin binding inhibitory immunoglobulin (r = 0.506, p < 0.001).
Conclusions:
This study demonstrates an association of HMGB1 and its receptors in the inflammatory mechanisms of GO. HMGB1, RAGE, and TLR2 blockers reduced the production of pro-inflammatory molecules, providing a rationale for blocking the HMGB1 pathway to treat patients with GO. HMGB1 proteins were secreted further in the plasma of patients with active GO, suggesting that HMGB1 can be used as a biomarker of GO activity.
Introduction
Graves' disease (GD) is an autoimmune disease that can be complicated by ocular manifestations localized to the orbit known as Graves' orbitopathy (GO). GO occurs in up to 50% of patients with GD and exhibits various clinical symptoms and signs (1). Orbital fibroblasts are the primary target cells in GO and secrete hyaluronan in response to various cytokines (1). Inflammation causes enlargement of the extraocular muscle bodies, soft-tissue swelling, and an overabundance of adipose tissue, which can result in proptosis, restrictive strabismus, and compressive optic neuropathy (2). Approximately 5–6% of patients with GO experience severe visual compromise due to extreme orbital inflammation (3). However, the precise pathological mechanism and inflammatory mediators contributing to the development and progression of GO remain unclear.
High-mobility group box 1 (HMGB1) is a highly conserved and ubiquitous non-histone protein containing two DNA binding domains, HMG box A and B, with an acidic C-terminal domain (4). HMGB1 is mainly located in the nucleus and interacts with DNA by regulating transcription, recombination, and repair. HMGB1 can be actively secreted by inflammatory immune cells (5) or passively released during apoptosis or necrosis (6). Outside of the cell, HMGB1 serves as an alarmin or damage-associated molecular pattern signal. Thus, HMGB1 functions as a cytokine to trigger responses by interacting with several receptors, including the receptor for advanced glycation end products (RAGE), Toll-like receptor (TLR) 2, and TLR4 (7). Extracellular HMGB1 stimulates the secretion of pro-inflammatory mediators and cytokines, which in turn promote HMGB1 secretion as a positive feedback loop during the inflammatory process (7).
Recent studies have indicated a crucial role for HMGB1 in autoimmune diseases and inflammatory diseases, such as systemic lupus erythematous (SLE), rheumatoid arthritis (RA), myositis, and autoimmune thyroiditis (8 –12). Increased HMGB1 expression has also been reported in adipose tissue in obese humans (13). As GO is an autoimmune inflammatory disease, it was hypothesized that HMGB1 participates in its pathogenesis. However, to the authors' knowledge, this potential association has not been examined to date.
This study investigated the involvement of HMGB1 in the inflammatory pathogenesis of GO in an in vitro model. The plasma levels of HMGB1 were compared in active or inactive GO patients, GD patients without GO, and healthy control subjects. Plasma HMGB1 levels were also correlated with the clinical activity score (CAS) and thyrotropin (TSH) receptor antibody (TRAb) levels. GO and non-GO orbital fibroblasts were treated with inflammatory cytokines to evaluate the effect of inflammation on HMGB1 and its receptors and their associated pathways. These findings may help to expand the association between HMGB1 and inflammatory autoimmune diseases, and reveal a potential of HMGB1 as a new biomarker and/or therapeutic target for treating GO.
Methods
Reagents
Recombinant human HMGB1 (rhHMGB1), recombinant human interleukin (IL)-1β, and tumor necrosis factor (TNF)-α were purchased from R&D Systems (Minneapolis, MN). Dulbecco's modified Eagle's medium/nutrient mixture F-12 (DMEM/F-12) was purchased from Lonza, Inc. (Basel, Switzerland). Fetal bovine serum (FBS) was purchased from Invitrogen (Carlsbad, CA). Penicillin-streptomycin was purchased from Welgene, Inc. (Gyeongsangbuk-do, Korea). Anti-HMGB1 antibodies were purchased from Abcam (Cambridge, United Kingdom). Anti-TLR2, anti-TLR4, and anti-beta actin antibodies were purchased from Santa Cruz Biotechnology (Dallas, TX). Anti-RAGE, anti-AKT (protein kinase B), anti-phosphorylated-AKT, anti-extracellular signal-related kinase (ERK), anti-phosphorylated-ERK, anti-Jun N-terminal kinase (JNK), anti-phosphorylated-JNK, anti-nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), and anti-phosphorylated-NF-κB (pNF-κB) antibodies were purchased from Cell Signaling Technology (Danvers, MA).
To inhibit HMGB1, RAGE, and TLR2, anti-HMGB1 blocking antibody, the RAGE antagonist (FPS-ZM1), and anti-TLR2 blocking antibody were purchased from Cell Signaling Technology, Merck KGaA (Darmstadt, Germany) and Santa Cruz Biotechnology, respectively. Enzyme-linked immunosorbent assay (ELISA) kits for the quantitative detection of HMGB1 in the human plasma and cell supernatant were purchased from Abbexa Ltd. (Cambridge, United Kingdom) and R&D Systems, respectively.
Subjects and preparation of tissues and cells
Orbital tissue specimens were collected from patients with GO during orbital decompression surgery (n = 6). Normal orbital tissue was obtained from age- and sex-matched control subjects with no history of autoimmune thyroid disease or GO during orbital surgery for other non-inflammatory diseases (n = 5). The protocol for obtaining orbital adipose/connective tissue and plasma from patients and healthy volunteers was approved by the Institutional Review Board of Severance Hospital, and written informed consent was obtained from all patients and healthy volunteers. This research adhered to the tenets of the Declaration of Helsinki. At the time of surgery, the GO patients were in a euthyroid state and had not been administered steroid or radiation therapy for at least three months.
For plasma HMGB1 evaluation, 175 subjects were recruited: 51 with active GO, 48 with inactive GO, 30 with GD without GO, and 46 healthy volunteers. CAS is based on the classical signs of inflammation and consists of seven items (14). The CAS ranges from 0 to 7, and GO is defined as “active” if the CAS is ≥3. Table 1 shows the demographic, clinical, and serologic data of the subjects.
Clinical and Serological Data of Patients and Controls for ELISA
ELISA, enzyme-linked immunosorbent assay; GO, Graves' orbitopathy; GD, Graves' disease; SD, standard deviation; IQR, interquartile ranges; T3, triiodothyronine; T4, thyroxine; TSH, thyrotropin; TBII, thyrotropin-binding inhibitory immunoglobulin; TSI, thyroid-stimulating immunoglobulin.
For the preparation of the orbital tissue study, orbital tissues were homogenized with a tissue homogenizer (Precellys® 24; Bertin Instruments, Montigny-le-Bretonneux, France) using a Precellys lysing kit (Bertin Instruments) with lysis buffer (150 mM sodium chloride, 1% Triton X-100, 1% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS), 50 mM Tris-HCl, pH 7.5, 2 mM EDTA, 1 mM AEBSF, 800 nM aprotinin, 50 μM bestatin, 15 μM E64, 20 μM leupeptin, and 10 μM pepstatin A). The protein contents were determined using a BCA protein concentration kit (Thermo Fisher Scientific, Waltham, MA).
For orbital fibroblast analysis, the specimens were primarily cultured as previously described (15). Briefly, minced tissue was suspended in DMEM/F-12 containing 20% FBS and penicillin-streptomycin. After the growth of fibroblasts, the cells were passaged with trypsin/EDTA. The cells were then incubated in DMEM containing 10% FBS and antibiotics. Cells from passages 2–5 were used for subsequent experiments. The orbital fibroblasts were grown to confluence in 6 cm dishes, and the culture medium was changed to serum-free DMEM/F-12, followed by incubation with 10 ng/mL of the inflammatory cytokine TNF-α or IL-1β.
Real-time PCR
Total RNA was isolated using TRIZOL (Invitrogen). One microgram of RNA was used for first-strand cDNA synthesis according to the manufacturer's instructions (#639543; Takara Bio, Inc., Shiga, Japan). mRNA levels were measured by RT-PCR with the ABI StepOnePlus real-time PCR system (Applied Biosystems, Foster City, CA) using SYBR green PCR master mix (Life Technologies, Carlsbad, CA).
The following primers were used: HMGB1 5′-GTT CAA GGA TCC CAA TGC AC-3′ (forward) and 5′-TGA TTT TTG GGC GAT ACT CAG-3′ (reverse); RAGE 5′-GCC ACT GGT GCT GAA GTG TA-3′ (forward) and 5′-TGG TCT CCT TTC CAT TCC TG-3′ (reverse); TLR2 5′-GAC TTC TCC CAT TTC CGT CT-3′ (forward) and 5′-CAG GTA GGT CTT GGT GTT CA-3′ (reverse); TLR4 5′-GAC CTT TCC AGC AAC AAG ATT C-3′ (forward) and 5′-GAG AGA TTG AGT AGG GGC ATT T-3′ (reverse); and GAPDH 5′-TGC TGT AGC CAA ATT CGT TG-3′ (forward) and 5′-CAC CCA CTC CTC CAC CTT T-3′ (reverse).
All PCR reactions were performed in triplicate, and the housekeeping gene GAPDH was used for normalization. The results are expressed as the fold change of the threshold cycle (Ct) value relative to that in the control group using the 2–ΔΔCt method (16).
Western blot assays
Equal amounts of protein (50 μg) were separated by 10% SDS polyacrylamide gel electrophoresis. The resolved proteins were transferred to nitrocellulose membranes and incubated overnight with primary antibodies at 4°C. Immunoreactive bands were detected using horseradish peroxidase–conjugated secondary antibodies. The membranes were then visualized with enhanced chemiluminescence solution and exposure to X-ray film (GE Healthcare, Piscataway, NJ) or an image reader (LAS-4000 mini; Fuji Photo Film, Tokyo, Japan). Densitometric quantification of each immunoreactive band was performed using Image J software (National Institutes of Health, Bethesda, MD), and values were normalized to the level of β-actin in the same sample.
ELISA for IL-6, IL-8, MCP-1, and HMGB1 detection in cell supernatant
IL-6, IL-8, and MCP-1 protein levels were measured in cell supernatants using a commercial ELISA kit according to the manufacturer's protocol. Secreted HMGB1 protein levels in cell supernatants were detected with a commercial ELISA kit according to the manufacturer's protocol.
Blood sampling and measurement of plasma HMGB1 and TRAb concentrations
Blood samples were drawn into test tubes containing a 1/10 volume of sodium citrate. Platelet-free plasma was obtained after centrifugation at 1500 g for 15 minutes at 4°C and stored at −80°C until analysis. Plasma HMGB1 protein levels were determined with a commercially available ELISA kit according to the manufacturer's protocol. All samples were tested in triplicate, and all sera were run in the same assay.
TRAb were measured with a third-generation TSH binding inhibitory immunoglobulin (TBII) assay using the automated Cobas electrochemiluminescence immunoassay (Elecsys; Roche Diagnostics GmbH, Penzberg, Germany) according to the manufacturer's instructions. Thyroid-stimulating immunoglobulin (TSI) in the patient serum was measured with the Thyretain™ TSI Reporter BioAssay (Diagnostic Hybrids, Inc., Athens, OH) according to the manufacturer's instructions.
Statistical analysis
IBM SPSS Statistics for Windows v20.0 (IBM Corp., Armonk, NY) was used for statistical analyses. All experiments were performed at least three times on samples from different patients, and the results are expressed as the mean values ± standard deviation. Comparisons of data between groups or within cell groups were analyzed with the t-test or one-way analysis of variance. The Mann–Whitney U-test and Kruskal–Wallis test were used for nonparametric data or not normally distributed with the Kolmogorov–Smirnov test. Spearman's rank correlation coefficient was applied to analyze the relationship of plasma HMGB1 titers with CAS and TRAb levels. A p-value of <0.05 denoted statistical significance.
Results
HMGB1 and its receptors are highly expressed in GO orbital tissues
To determine the basal mRNA expression levels of HMGB1 and its receptors in GO and non-GO orbital tissues, orbital tissue explants were obtained from GO (n = 6) and non-GO (n = 5) control subjects, and relative quantification was performed by RT-PCR. The mean values of 2–ΔΔCt values from RT-PCR data of HMGB1, RAGE, TLR2, and TLR4 mRNAs were significantly higher in GO orbital tissues than in non-GO tissues (Fig. 1). The mRNA levels were consistently low in all non-GO tissues.
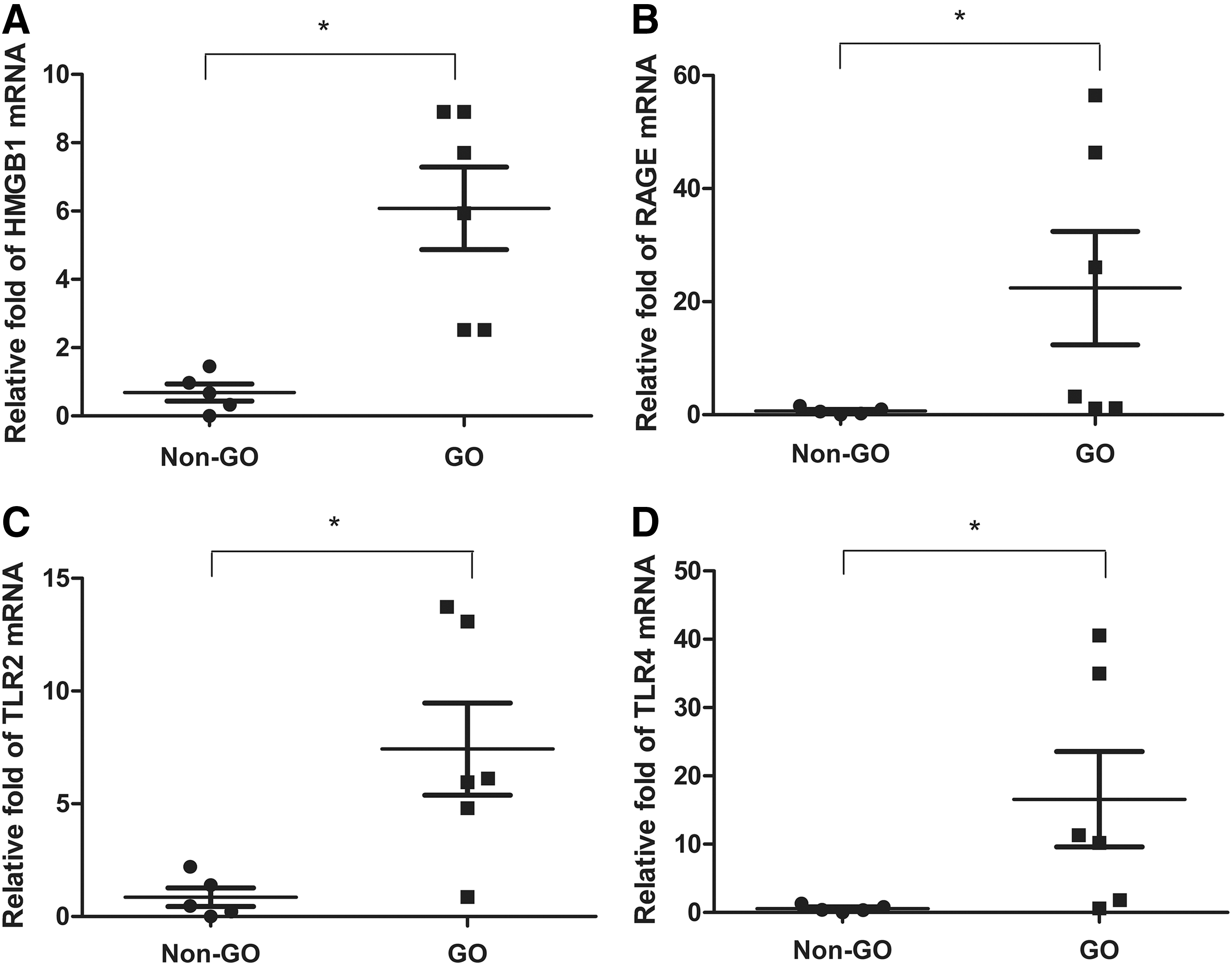
mRNA transcript levels of high-mobility group box 1 (HMGB1) and its receptors in Graves' orbitopathy (GO) and non-GO orbital tissues. Orbital tissues were obtained from six GO patients and five non-GO control subjects, and the samples were assayed in triplicate. The mRNA expression of each HMGB1 (
HMGB1 protein expression was analyzed in GO (n = 4) and non-GO (n = 4) orbital tissue lysates by Western blotting and densitometry scanning. HMGB1 protein expression was significantly higher in GO tissues than in non-GO tissues (Fig. 2).
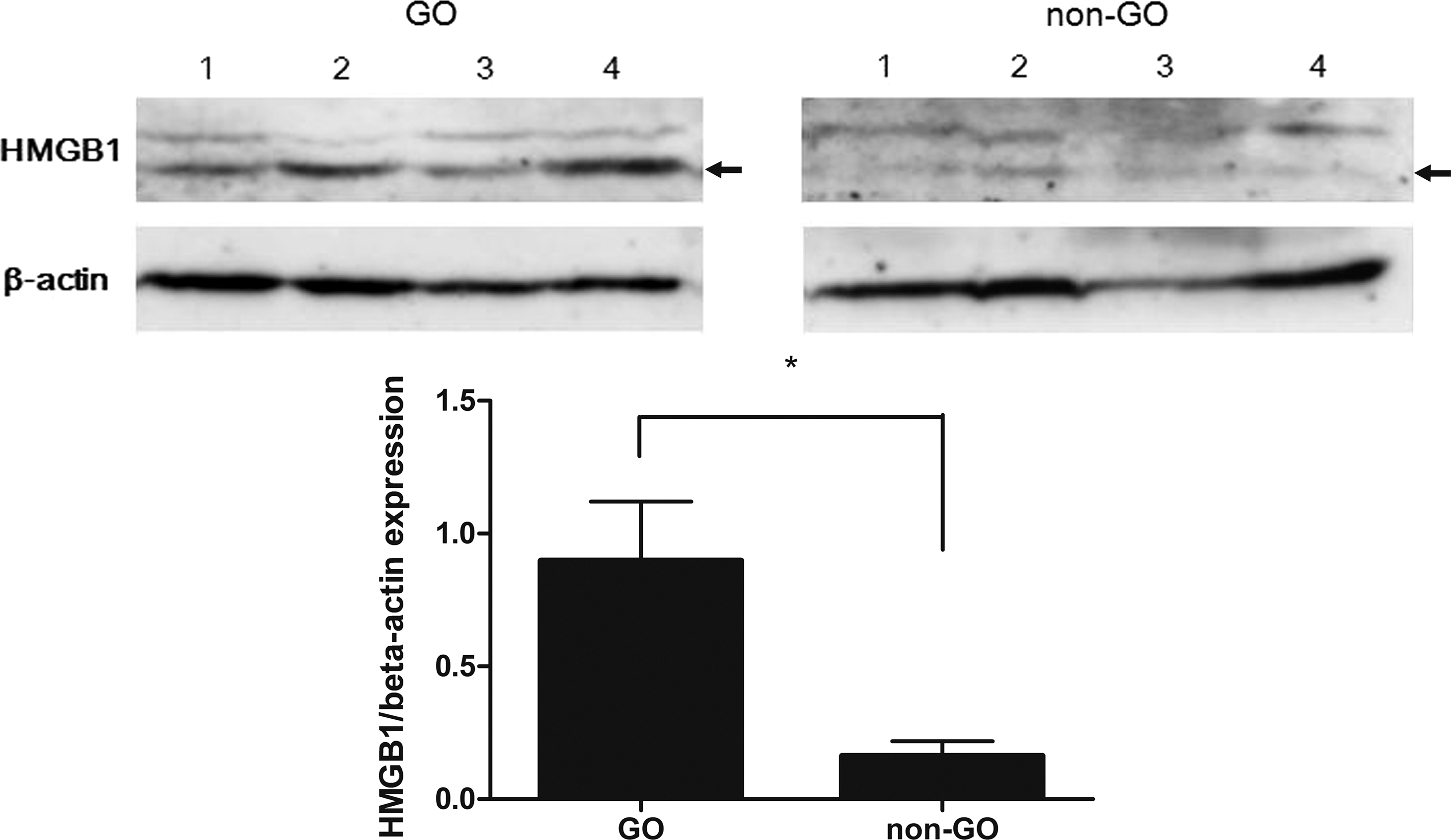
Expression of HMGB1 protein in GO and non-GO tissues. HMGB1 protein expression was assayed by Western blot analysis in four GO and four non-GO tissues isolated from different patient samples, and the samples were assayed in triplicate. Representative blots and quantification of HMBG1 by densitometry, normalized to the level of β-actin in the same sample, are shown. The data in the columns are mean relative density/β-actin fold relative to GO ± standard deviation (SD). Differences between GO and non-GO tissues are indicated (*p < 0.05). Arrows indicate 29 kDa.
mRNA levels of HMGB1 and its receptors are highly expressed in GO orbital fibroblasts under inflammatory conditions
The study investigated whether pro-inflammatory cytokines such as IL-1β and TNF-α induce HMGB1 and its receptors in primary cultured orbital fibroblasts. The mRNA transcript levels of HMGB1 and its receptors under inflammatory stimulation in both GO (n = 3) and non-GO (n = 3) orbital fibroblasts were evaluated by RT-PCR. HMGB1, RAGE, and TLR2 mRNA expression levels were markedly increased in orbital fibroblasts from GO patients (black columns in Fig. 3) after stimulation with IL-1β (Fig. 3A) and TNF-α (Fig. 3B).

Effect of interleukin (IL)-1β and tumor necrosis factor (TNF)-α treatment on mRNA expression of HMGB1 and its receptors. Orbital fibroblasts from subjects with GO (black column, n = 3) and without GO (white column, n = 3) were stimulated with 10 ng/mL (
HMGB1 protein secretions are increased under inflammatory conditions
GO (n = 3) and non-GO cells (n = 3) were stimulated with 10 ng/mL IL-1β or TNF-α for various time periods (0–48 hours), and the secretions of HMGB1 protein were determined by ELISA (Fig. 4). Protein secretion of HMGB1 was increased in a time-dependent manner following stimulation with IL-1β (Fig. 4A) or TNF-α (Fig. 4B) in both GO and non-GO cells, and enhanced HMGB1 secretion was significantly higher in GO cells (black column) than in non-GO cells (white column).
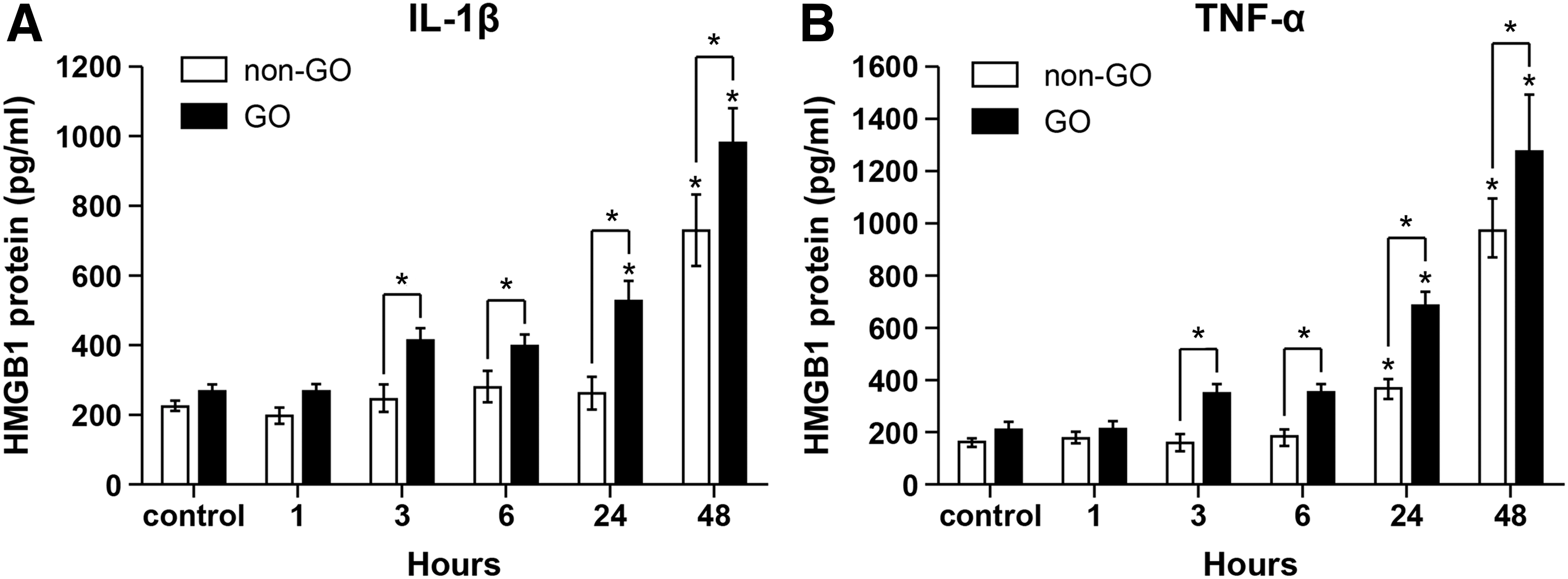
HMGB1 protein secretion under inflammatory conditions in GO and non-GO cells. GO (black column, n = 3) and non-GO (white column, n = 3) cells were stimulated with 10 ng/mL of IL-1β (
Exogenous recombinant HMGB1 upregulates RAGE and TLR2 expression
Following treatment of GO orbital fibroblasts with 200 ng/mL of rhHMGB1 for 0–24 hours, RAGE and TLR2 protein expression levels were significantly increased, as determined by Western blot analysis. RAGE protein expression immediately increased one hour after rhHMGB1 stimulation, reached a maximal level at three hours, and returned to baseline levels after 24 hours of treatment. TLR2 protein levels were markedly increased at 16 hours after rhHMGB1 treatment. In contrasts, TLR4 protein levels were not influenced by rhHMGB1 stimulation (Supplementary Fig. S1).
Blockade of HMGB1, RAGE, and TLR2 suppressed phosphorylation of NF-κB and pro-inflammatory cytokines production under inflammatory conditions in GO
To evaluate whether HMGB1 and its receptors are involved in the NF-κB signaling pathway, GO orbital fibroblasts were pretreated with an anti-HMGB1 antibody (1 μg/mL), FPS-ZM1 (200 ng/mL), or anti-TLR2 antibody (1 μg/mL) for 30 minutes before IL-1β or TNF-α stimulation for 24 hours (Fig. 5). Phospho-NF-κB protein expression was increased after stimulation with IL-1β or TNF-α, and significantly attenuated by treatment with anti-HMGB1 antibody (Fig. 5A), FPS-ZM1 (Fig. 5B), and anti-TLR2 antibody (Fig. 5C).
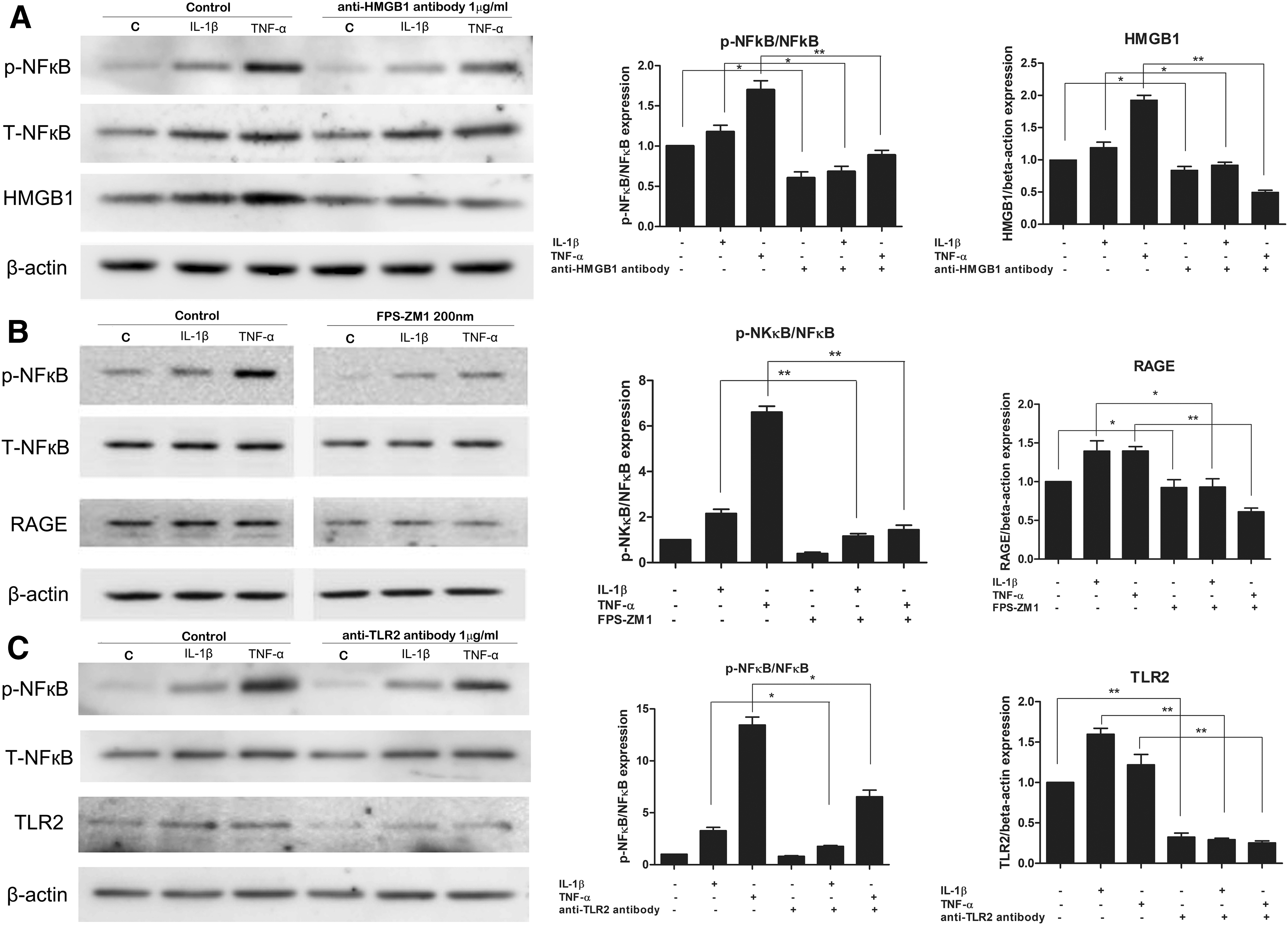
Effect of HMGB1, RAGE, and TLR2 inhibitors on the phosphorylation of anti-nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB). Cells from GO patients (n = 3) were pretreated with anti-HMGB1 antibody (1 μg/mL) (
To determine whether HMGB1 and its receptors are involved in the synthesis of pro-inflammatory cytokines, GO cells were pretreated with the anti-HMGB1 antibody (1 μg/mL), FPS-ZM1 (200 ng/mL), or anti-TLR2 antibody (1 μg/mL) for 30 minutes before IL-1β or TNF-α stimulation for 24 hours (Fig. 6). IL-1β or TNF-α induced production of IL-6 (Fig. 6A), IL-8 (Fig. 6B), and MCP-1 (Fig. 6C), and they were significantly reduced by anti-TLR2 antibody. Treatment with an anti-HMGB1 antibody also inhibited the secretion of pro-inflammatory cytokines, except for IL-1β, which induced MCP-1 production. FPS-ZM1 significantly reduced the secretion of unstimulated IL-6 protein and IL-1β induced IL-8 protein. However, it did not change the release of MCP-1 protein.
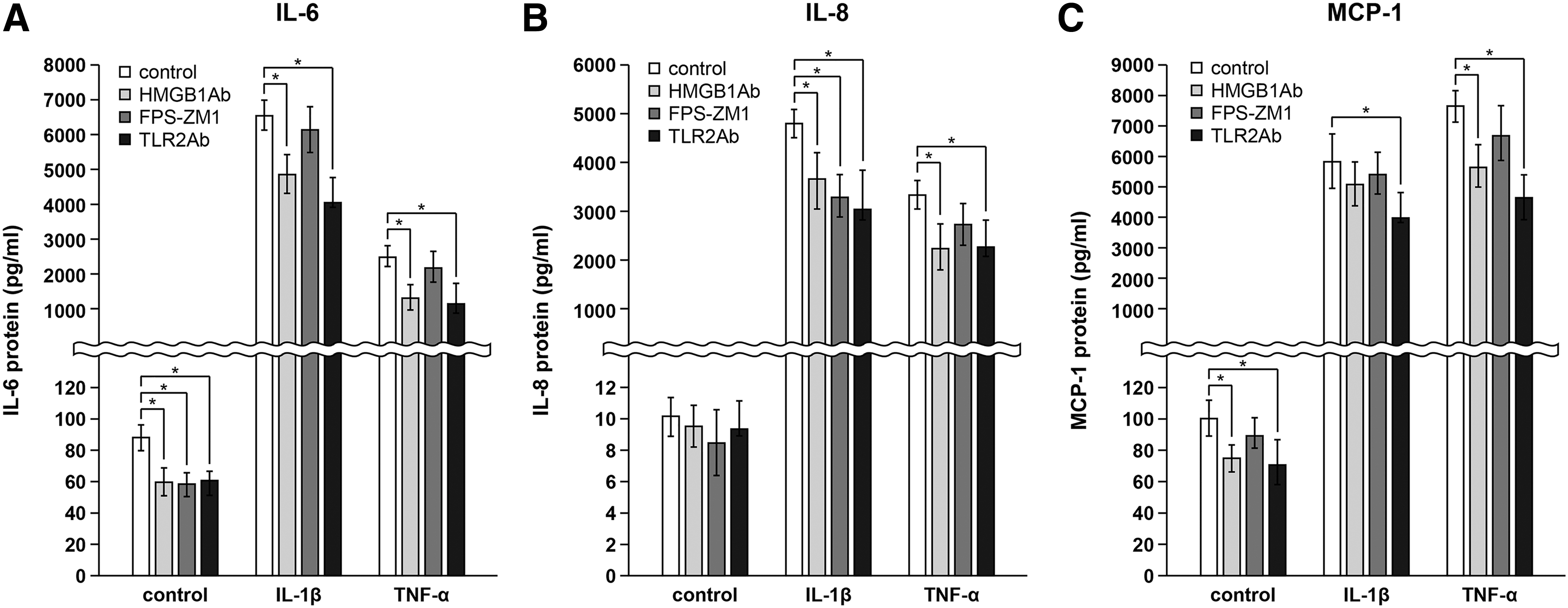
Blockade of HMGB1, RAGE, and TLR2 on the production of pro-inflammatory cytokines. GO cells were pretreated with anti-HMGB1 antibody (1 μg/mL; light-gray column), FPS-ZM1 (200 ng/mL; dark-gray column), or anti-TLR2 antibody (1 μg/mL; black column) for 30 minutes and stimulated with 10 ng/mL IL-1β or TNF-α for 24 hours. Release of IL-6 (
HMGB1 activates ERK, JNK, and AKT signaling pathways
The expression levels of ERK, JNK, AKT, and their phosphorylated forms were analyzed by Western blot analysis (Fig. 7). Treatment of orbital fibroblasts with 100 ng/mL rhHMGB1 rapidly activated ERK, whose phosphorylation reached a maximum at 15 minutes in both GO (Fig. 7A) and non-GO orbital fibroblasts (Fig. 7B) and then decreased to baseline levels at 180 minutes. JNK phosphorylation was rapidly and strongly enhanced in GO and non-GO cells, with maximum levels detected at 5 and 15 minutes, respectively. AKT phosphorylation was enhanced only in GO cells, with a maximum level detected at 60 minutes.
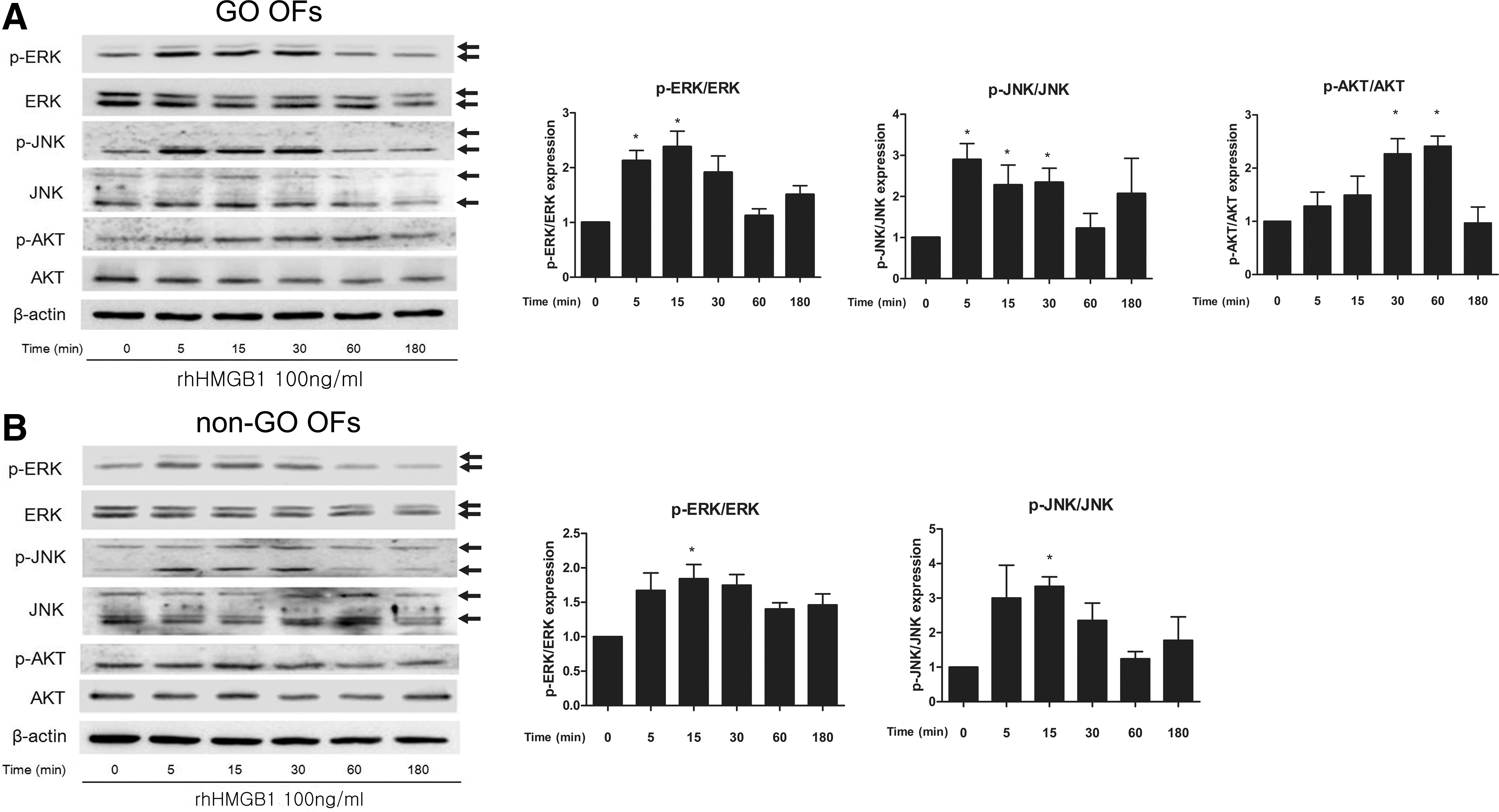
Effect of exogenous recombinant HMGB1 on signaling proteins in GO and non-GO cells. Phosphorylation of ERK, JNK, and AKT was investigated by Western blot analysis in orbital fibroblasts of (
Plasma HMGB1 protein levels are increased in active GO
Plasma HMGB1 levels were measured in patients with active/non-active GO, GD without GO, and healthy subjects by ELISA (Fig. 8). The mean plasma level of HMGB1 in patients with active GO (n = 51, M ± SD = 4107.56 ± 1246.01 pg/mL [range 2354–7664 pg/mL]) was significantly higher than those in patients with inactive GO (n = 48, M ± SD = 2458.58 ± 881.73 pg/mL [range 1240–5154 pg/mL]; p < 0.001), GD patients without GO (n = 30, M ± SD = 2043.00 ± 536.11 pg/mL [range 763–2584 pg/mL]; p

Comparison of plasma levels of HMGB1 among patients with active GO, inactive GO, and GD without GO, and healthy control subjects. Plasma HMGB1 levels were measured in patients with active/non-active GO and GD without GO, and healthy subjects by ELISA. The samples were assayed in triplicate. The mean HMGB1 level was significantly higher in the plasma of active GO (n = 51) than in the plasma of inactive GO (n = 48), GD without GO (n = 30), and healthy subjects (n = 46). The results of the data are presented as the medians and interquartile ranges of values determined by ELISA (*p < 0.001 vs. each group).
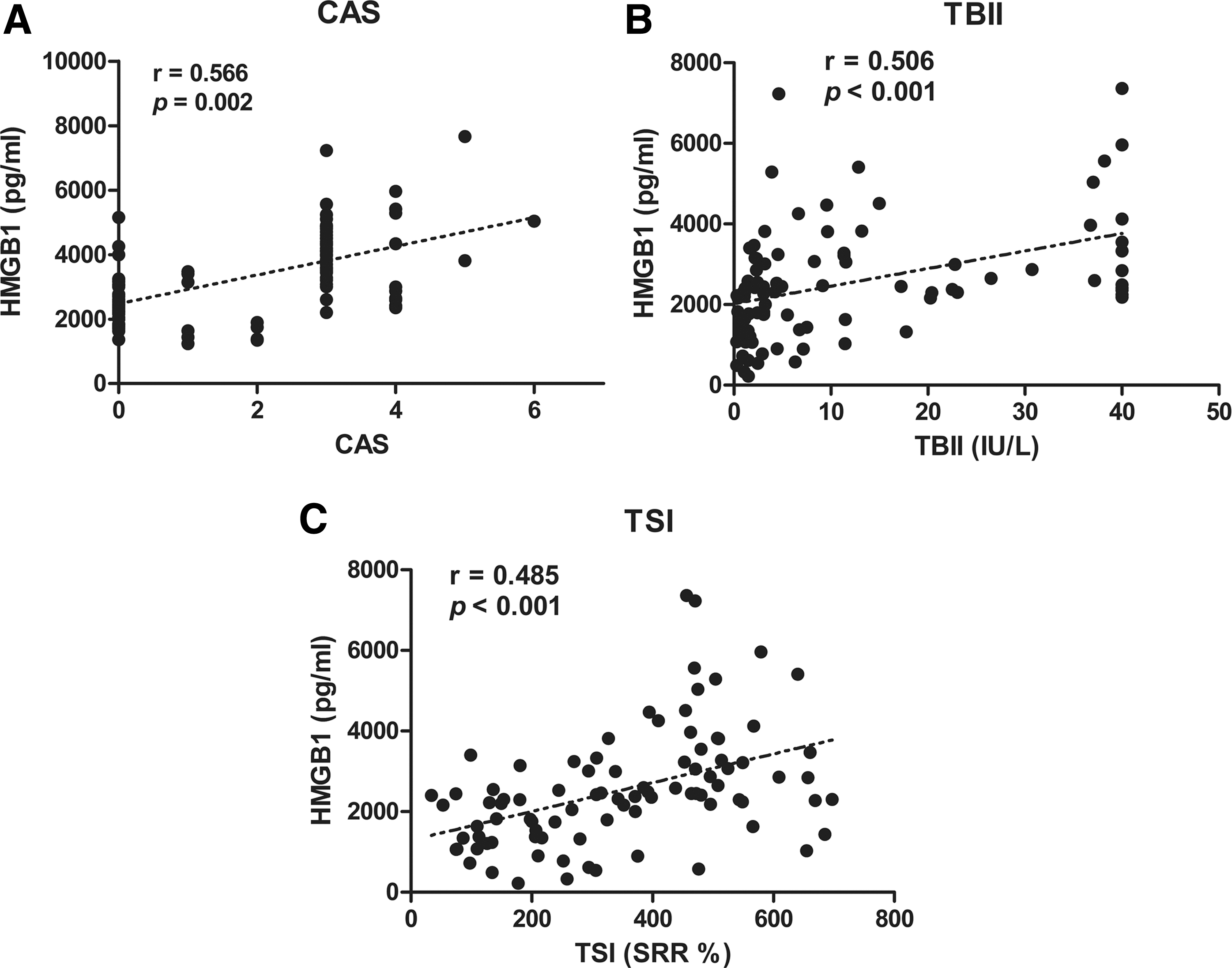
Correlation analyses between plasma levels of HMGB1 and clinical activity score (CAS) or thyrotropin receptor antibody. Correlation between plasma levels of HMGB1 and (
Discussion
This study investigated whether HMGB1 is linked to the inflammatory pathogenesis of GO, as one of the main mechanisms of orbital change in GO is associated with autoimmune inflammation. Although the precise etiology of GO remains unknown, it has been shown to involve the infiltration of activated T cells producing pro-inflammatory cytokines that induce glycosaminoglycan and hyaluronan accumulation, along with orbital adipogenesis, resulting in orbital fibrosis and edema (1). T and B lymphocytes, inflammatory cells, macrophages, and mast cells interact with GO orbital fibroblasts through a sequence of cytokines, which magnify the activation of orbital fibroblasts (1). Moreover, a previous study showed that the mRNA and protein levels of IL-1β and TNF-α were significantly increased in GO tissues compared to non-GO tissues (17).
Supporting the hypothesis that HMGB1 is related to GO inflammation, first it was observed that the mRNA and protein levels of HMGB1 were higher in GO tissue explants than in non-GO tissues. Gene levels of RAGE, TLR2, and TLR4 were also significantly higher in GO tissue explants than in non-GO tissues. In addition, HMGB1, RAGE, and TLR2 mRNA were increased after stimulation with IL-1β or TNF-α—to a greater extent in GO fibroblasts than in non-GO fibroblasts. We also identified that HMGB1 secretion was further increased after stimulation with IL-1β or TNF-α in GO cells than in non-GO cells.
The specific mechanism of HMBG1 involvement in GO pathogenesis remains unclear. However, it was predicted that HMGB1 is an important effector molecule in the milieu of local inflammation of GO, as it has been implicated in other diseases, such as RA and SLE. The number of HMGB1-secreting cells and levels of HMGB1 were increased at distinct sites of inflammation, including the inflamed synovium of RA patients, and anti-HMGB1 antibodies blocked the development of synovial inflammation in an experimental arthritis model (10,18). HMGB1 is actively secreted by immune cells or tissue under inflammatory or hypoxic conditions (6), and HMGB1 may induce cytokines, promote chemotaxis, activate cells such as immune cells, endothelial cells, and fibroblasts, or stimulate the production of autoantibodies as previously reported (7 –13,19). HMGB1 induces human monocytes to release various inflammatory cytokines, including IL-6, IL-8, TNF-α, IL-1α, IL-1β, MIP-1α, and MIP-1β (20). HMGB1 acquires enhanced pro-inflammatory activity after incubation with IL-1β through its role as a carrier protein for cytokines (21). In this regard, HMGB1 liberation from orbital fibroblasts stimulated with cytokines such as IL-1β or TNF-α may boost cellular interactions, including T cells, B cells, and macrophages, but further experiments are required to clarify the mechanism. GO is associated with the presence of autoantibodies against thyroidal antigens, including TRAb. TBII along with a TSI bioassay are both helpful for diagnosis and prognostic prediction in GO (22). The current study shows that plasma HMGB1 levels in patients with active GO were significantly higher than in patients with inactive GO, or patients with GD without GO, as well as in healthy subjects. It has also been found that plasma levels of HMGB1 revealed a highly significant correlation with the level of TRAb, a well-known inflammatory activity indicator and an important pathogenic contributor to GO (23), as well as CAS. Elevated HMGB1 levels have been reported not only in diseased tissue, but also in the blood and other biological fluids, such as synovial fluid and urine in various autoimmune diseases (24 –26). The HMGB1 concentration was positively correlated with the disease activity of RA (27), SLE (24), and antineutrophilic cytoplasmatic antibody-associated vasculitis (28). It is expected that HMGB1 may play a diagnostic and predictive role of HMGB1 in the GO field in the future.
TLRs have been shown to mediate the activation of NF-κB and mitogen-activated proteins kinase, resulting in the production of mediators of the innate immune system, such as IL-1, IL-6, IL-8, or TNF-α (29 –31). TLR2 and TLR4 both play crucial roles in the development of autoimmune diseases, and activation of both TLRs can upregulate inflammatory cytokines and chemokines (29). The expression levels of TLR2 and TLR4 were demonstrated to be higher on synovial fibroblasts and macrophages in RA patients than in osteoarthritis patients and healthy donors (32). RAGE is a multiligand receptor that binds to HMGB1, S100 family members, and amyloid-β (33,34). RAGE also functions as an endothelial adhesion receptor for leukocyte integrins and promotes leukocyte recruitment (35). Blockade of RAGE suppresses inflammation and reduces tissue damage in experimental models of inflammatory disorders, thus identifying RAGE as a possible therapeutic target in inflammation (36). An HMGB1 antagonist suppressed inflammatory infiltration in an autoimmune thyroiditis mouse model by inhibiting TLR2-HMGB1 pathways (12). TLR4 gene polymorphisms did not differ between GD patients with and without GO (37). The expression level of TLR2 mRNA, but not TLR4 mRNA, in peripheral blood mononuclear cell samples was significantly increased in GD subjects compared to healthy controls, and increased IL-6 and decreased IL-10 production in these cells from autoimmune thyroid disease patients was related to TLR2 (38). In this study, RAGE and TLR2 appeared to mediate HMGB1 response in orbital fibroblasts. The reasons why RAGE or TLR2, but not TLR4, was involved in GO remain unclear. IL-1β or TNF-α induced production of IL-6, IL-8, and MCP-1, and phosphorylation of NF-κB was suppressed by treatment with an anti-HMGB1 antibody, an anti-TLR2 antibody, and a RAGE antagonist. The suppressive effect of all inhibitors was not uniform, probably because of the complexity of the HMGB1 pathway in the inflammatory mechanism. Nevertheless, consistent with other studies on other inflammatory disease, this study found that blockage of HMGB1, RAGE or the TLR2 receptor pathway could be a new potential therapeutic target for managing GO.
This study demonstrates that phosphorylated ERK, JNK, and AKT proteins were upregulated by rhHMGB1 treatment in GO orbital fibroblasts, and phosphorylation of AKT by HMGB1 stimulation was only detected in GO cells. Previous studies indicated that AKT phosphorylation is increased by TSH and insulin-like growth factor 1 stimulation (39), which are major pathogenic stimuli in GO (1), in GO orbital fibroblasts. Adipocyte differentiation and adipogenic markers such as adiponectin and peroxisome proliferator-activated receptor γ were reduced by treatment with the phosphoinositide 3-kinase (PI3K) inhibitor LY294002 (40,41). It was recently reported that Idelalisib, a selective PI3Kδ inhibitor, not only reduced AKT phosphorylation, but also inhibited IL-1β-induced expression of IL-6 and IL-8 (42). It would be interesting to perform further in vitro studies to modulate GO pathogenesis by targeting the HMGB1-AKT signaling pathway.
Human primary orbital fibroblast cultures obtained from patients with GO and normal subjects were evaluated. All six GO patients presented signs of minimal inflammatory activity and were in a euthyroid state at the time of surgery. As orbital decompression surgery is mostly performed during the inactive stage of GO, the GO cell strains used were not in a highly active inflammatory condition, which could explain similar upregulations of proinflammatory cytokine proteins induced by IL-1β or TNF-α in both GO and non-GO cells, in agreement with previous studies (15,43). Cultures obtained from inflamed orbits with a CAS of ≥3 might respond differently compared to those from stable, inactive orbits. However, the gene and protein levels of HMGB1 and its receptors were different between GO and normal cells. Although clinical inflammatory states were not severe at the time of harvest, the enlarged, compressed adipose connective tissue in the fixed bony orbits of GO patients could have been subject to heightened, local inflammation compared to normal orbital fatty tissues.
This study is the first report of the pro-inflammatory activity of HMGB1 in GO orbital fibroblasts. The results suggest HMGB1 is related to the inflammatory activities of GO pathogenesis through binding to the RAGE and TLR2 receptors. The results show that HMGB1 secretion in response to pro-inflammatory cytokines, IL-1β or TNF-α, was greater in GO cells in comparison to non-GO cells. It was found that production of pro-inflammatory cytokines was suppressed by inhibition of the HMGB1 signaling pathway. The study also demonstrates that HMGB1 is associated with the NF-κB signaling pathway and activation of phosphorylated ERK, JNK, and AKT. The current study shows that plasma HMGB1 levels were significantly higher in patients with active GO when compared to those in patients with inactive GO, patients with GD without GO, and healthy subjects. The positive correlations of plasma HMGB1 with CAS and both TBII and TSI suggest the possibility of HMGB1 as a biomarker for active inflammation in GO patients. Overall, the results suggest that HMGB1 and its related signaling pathways may be potential targets for developing new therapeutics to treat inflammation associated with GO.
Footnotes
Acknowledgments
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (NRF-2017R1A2B4009565).
Author Disclosure Statement
The authors declare that they have no conflicts of interest.
Supplementary Material
Supplementary Figure S1