
Abstract
The co-occurrence of resistance to thyroid hormone beta (RTHβ) and myotonic dystrophy type 1 (DM1) was observed in a Japanese family. Two mutations, P453A and C36Y, were identified in the thyroid hormone receptor beta (THRB) gene. Whereas family members with THRBP453A exhibited RTHβ, two members with THRBC36Y but without THRBP453A had normal thyroid function. Two members, one with RTHβ and the other without, had a triplet expansion in the dystrophia myotonia protein kinase gene, a hallmark of DM1. The member with both RTHβ and DM1 developed atrial fibrillation at the age of 16 years, suggesting a synergistic impact on the heart.
Resistance to thyroid hormone beta (RTHβ), caused by mutations in the thyroid hormone receptor beta (THRB) gene, is a syndrome characterized by reduced responsiveness of the peripheral tissues to thyroid hormone. Tachycardia is a frequently observed symptom in subjects with RTHβ (1,2). Myotonic dystrophy type 1 (DM1) is an autosomal dominant disorder caused by the expansion of CTG triplets in the 3′-untranslated region of the dystrophia myotonia protein kinase (DMPK) gene, and patients often suffer from cardiac conduction defects in addition to myotonia, as well as progressive weakness and wasting (3). No patient with both RTHβ and DM1 has been previously reported.
A 47-year-old Japanese woman (the proband, I-2; Fig. 1) presented at the health-care center of the authors' hospital with moderate goiter, and thyroid function tests revealed an increase in thyroxine combined with an unsuppressed thyrotropin (TSH). Her past medical record revealed a diagnosis of hyperthyroxinemia at the age of 20 years. A sequence analysis of the THRB gene revealed two mutations: a substitution of cysteine by tyrosine at codon 36 in exon 4 (C36Y, c. 107 G>A), and of proline by alanine at codon 453 in exon 10 (P453A, c. 1357 c>G). The patient had three children. Her son (II-3) was healthy, had normal thyroid function tests, and carried the THRBC36Y but not the THRBP453A mutation. Her younger daughter (II-2) had moderate goiter and increases in free triiodothyronine and thyroxine with an unsuppressed TSH. A sequence analysis of individual II-2 identified the THRBP453A but not the THRBC36Y alteration. The index patient had been diagnosed as having DM1 at 14 years of age, and Southern blotting demonstrated an expansion of CTG repeats in the DMPK gene (400–550 repeats). The elder daughter (II-1) had normal thyroid function and carried THRBC36Y but not THRBP453A . She also had been diagnosed as having DM1, with an expansion of CTG repeats in DMPK gene (800–1300 repeats).
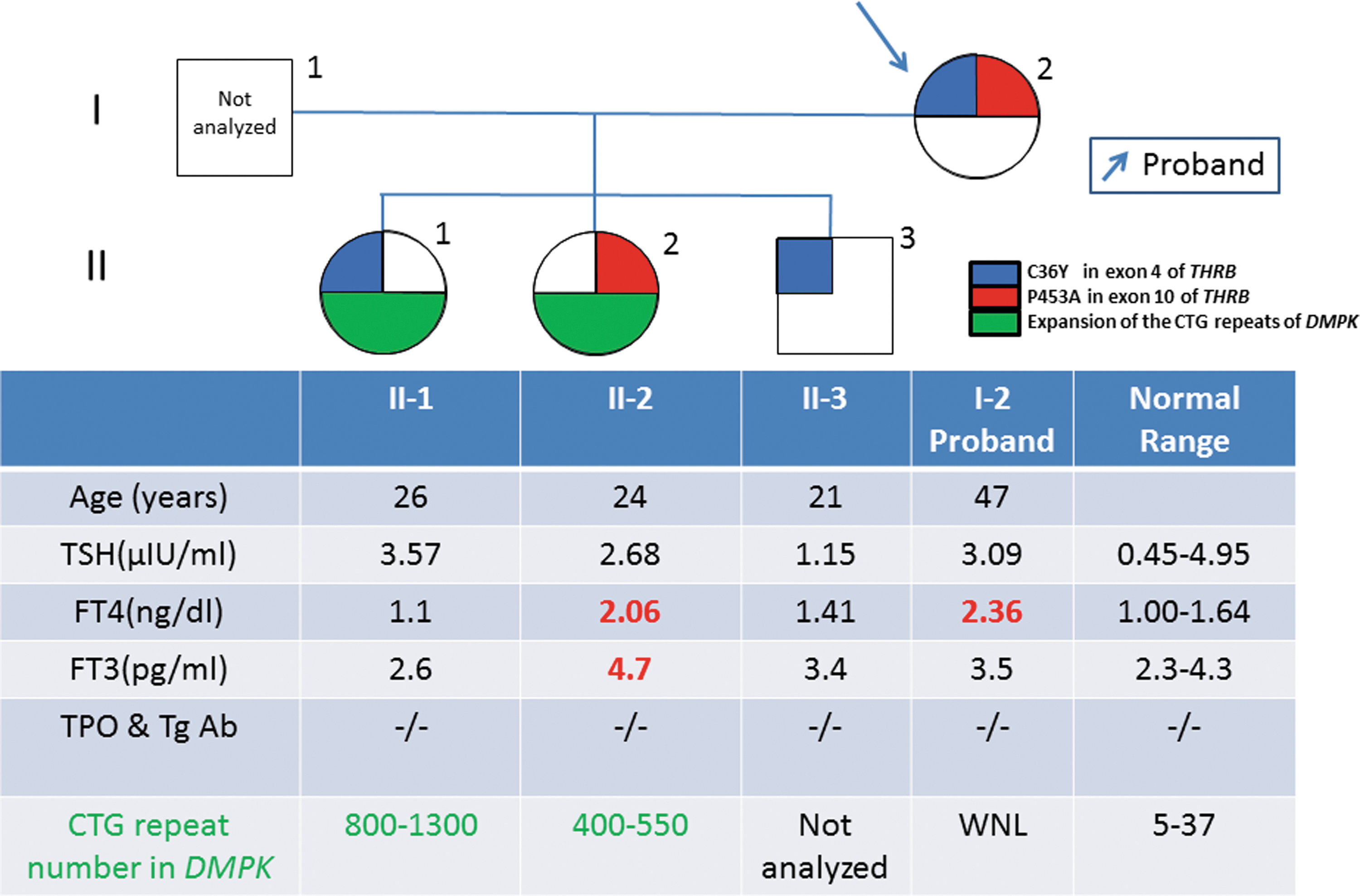
Thyroid function tests and genetic analysis results. Serum thyrotropin, free thyroxine, and free triiodothyronine levels were measured using Elecsys 2010 (Roche Diagnostics GmbH, Penzberg, Germany), and thyroid peroxidase antibody and thyroglobulin antibody titers were measured using Elecsys A-Ag and A-TPO (Roche Diagnostics GmbH), respectively. The two mutations in THRB—C36Y in exon 4 and P453A in exon 10—are shown in blue and red, respectively. The green indicates the expansion of the CTG repeats of the dystrophia myotonia protein kinase gene. Color images are available online.
In the present family, THRBP453A and THRBC36Y were transmitted independently from the proband to the children, indicating that they occur on different alleles. Since the thyroid function tests in II-1 and II-3 were normal, THRBC36Y does not produce the phenotype of RTHβ in a dominant manner. Furthermore, THRBC36Y has been registered in a single nucleotide polymorphism database as rs758677446. According to a genome aggregation database, the THRBC36Y variant is specific to East Asian populations, with an allele frequency of approximately 0.1%. On the other hand, THRBP453A has been identified in a number of families with RTHβ, and the proband is most likely a compound heterozygote for THRBP453A and THRBC36Y . As the thyroid function tests of the proband were comparable to II-2, who carries THRBP453A but not THRBC36Y , the C36Y mutation is unlikely to attenuate the function of THRB to a large degree. Indeed, an in silico PolyPhen-2 algorithm predicted that the effect of C36Y on THRB function would be benign, with a score of zero. However, it should be noted that THRB1 but not THRB2, which is predominantly expressed in the pituitary, contains the region encoded by exon 4. Therefore, C36Y in the N-terminal A/B domain is a THRB1-specific mutation.
In DM1, mutant RNAs containing expanded CUG repeats sequester proteins involved in the regulation of alternative splicing, leading to cardiac dysfunctions (4). Supraventricular arrhythmias are observed in 80% of patients during the later stage of disease, and the size of the CTG expansion is associated with the total number of deaths and cardiac dysfunctions, as well as the severity of progressive myopathy, including multiple organ disturbances. In the present family, II-2, who exhibited both RTHβ and DM1 and had a typical myopathic face, a highly myotonic gait, and alopecia, developed atrial fibrillation at 16 years of age. On the other hand, no cardiac defects or myopathic symptoms except for an elevated serum creatine kinase level have so far been apparent in the older daughter (II-1) who is 32 years of age, although she was found to have a further expansion of the CTG repeats compared to those present in II-2. The proband (I-2) experienced atrial flutter at 43 years of age and has been treated with β-blockade to control sustained tachycardia. The early onset of myopathic symptoms and atrial fibrillation in II-2 may have been caused by the combined deleterious effects of both disorders on skeletal and cardiac muscles that predominantly express THR alpha (THRA). The elevated thyroid hormone levels in II-2 may have stimulated myofibrillar proteolysis, resulting in acceleration of the muscular degeneration. To the authors' knowledge, this is the first report of coexistent RTHβ and DM1 mutations in an individual. Evaluation of patients who have hyperthyroidism in addition to DM1 in the future may clarify whether excessive thyroid hormone action in muscle aggravates the myopathy associated with DM1.
Footnotes
Author Disclosure Statement
No competing financial interests exist.