
Abstract
Background:
The thyrotropin receptor (TSHR) is the target for autoimmune thyroid stimulating antibodies (TSAb) triggering hyperthyroidism. Whereas elevated thyroid hormone synthesis by the thyroid in Graves' disease can be treated by antithyroid agents, for the pathogenic activation of TSHR in retro-orbital fibroblasts of the eye, leading to Graves' orbitopathy (GO), no causal TSHR directed therapy is available.
Methods:
Due to the therapeutic gap for severe GO, TSHR inhibitors were identified by high-throughput screening in Chinese hamster ovary cells expressing the TSHR. Stereo-selective synthesis of the screening hits led to the molecule S37, which contains seven chiral centers. Enantiomeric separation of the molecule S37 resulted in the enantiopure molecule S37a—a micro-molar antagonist of thyrotropin-induced cyclic adenosine monophosphate accumulation in HEK 293 cells expressing the TSHR.
Results:
The unique rigid bent shape of molecule S37a may mediate the observed high TSHR selectivity. Most importantly, the closely related follitropin and lutropin receptors were not affected by this compound. S37a not only inhibits the TSHR activation by thyrotropin itself but also activation by monoclonal TSAb M22 (human), KSAb1 (murine), and the allosteric small-molecule agonist C2. Disease-related ex vivo studies in HEK 293 cells expressing the TSHR showed that S37a also inhibits cyclic adenosine monophosphate formation by oligoclonal TSAb, which are highly enriched in GO patients' sera. Initial in vivo pharmacokinetic studies revealed no toxicity of S37a and a remarkable 53% oral bioavailability in mice.
Conclusion:
In summary, a novel highly selective inhibitor for the TSHR is presented, which has promising potential for further development for the treatment of GO.
Introduction
T
Autoantibodies mimic the natural ligand thyrotropin (TSH) by binding to the TSHR at the identical extracellular site as TSH, and cause deregulated TSHR hyperactivation (2). This pathogenic activation of the TSHR leads to uncontrolled production of the thyroid hormones thyroxine and triiodothyronine, causing hyperthyroidism like in Graves' disease (GD) (3).
Up to 80–90% of patients with hyperthyroidism suffer from GD, an autoimmune disease. To date, TSHR hyperactivation is treated by antithyroid drugs correcting the “clinically evident” consequences by pharmacological downstream inhibition of thyroid hormone synthesis in the thyroid gland.
Thus, the current primary antithyroid treatment does not target the causative molecular activation of the TSHR by antibodies, and patients are therefore burdened by adverse effects at a rate of at least 5% (4). In contrast to these drugs, which regulate thyroid hormone levels, the most promising target may be the TSHR itself.
Therapeutic gap for Graves' orbitopathy
The TSHR is expressed mainly in follicular epithelial cells of the thyroid gland, but also in retro-orbital fibroblasts (5). About 25% of GD patients develop an orbitopathy referred to as Graves' orbitopathy (GO), a related organ-specific autoimmune disease affecting the structure and function of the eyes (6 –9). There is considerable evidence that expression of the TSHR in the orbital fibroblasts (OF) and orbital adipocytes behind the eye contribute to this disease, which can pose a therapeutic challenge (10,11). OF have been recognized as primary target cells of autoimmune attack, and the TSHR (12) also acts as a primary auto-antigen in GO. The pathogenic activation of the TSHR by autoantibodies in retro-ocular fibroblasts initiates distinct signaling pathways compared to the thyroid (13,14). Apart from cyclic adenosine monophosphate (cAMP) activation (15), it leads to the production of extracellular matrix by the involvement of hyaluronan (HA) (15,16), fibrosis, and swelling of the extraocular muscle (10), as well as adipogenesis of OF (orbital fat expansion, compression of the optic nerve, and exophthalmos) (12,14,17 –19). Human retro-ocular fibroblasts have been utilized as in vitro models to study GO (20). Although in >60% of the cases GO ameliorates when thyroid function is normalized, the severity of the disease remains stable or even progresses in the remaining patients (21).
In addition, the current form of GD treatment is often inefficient because (i) in the majority of patients, antithyroid drug treatment does not lead to long-term euthyroidism, and consequently the thyroid gland must be removed by surgery or radioiodine ablation; and (ii) this ablative treatment is associated with risks such as recurrent nerve damage and hypoparathyroidism after surgery. Current treatment of severe GO forms (22) is largely imperfect and associated with a therapeutic dilemma (23).
A pharmacological approach to circumvent these obstacles is the suppression of pathogenic TSHR activation by drugs such as orally applicable TSHR-specific allosteric small-molecule antagonists (SMA) (24,25), which are not yet available clinically.
Pharmacological approaches to tackle GO
SMA have a strong therapeutic potential for TSHR-mediated GO. In contrast to the activating autoantibodies that bind like TSH to the extracellular region of the TSHR (26), synthetic SMA preferentially bind elsewhere, for example allosterically into a binding pocket located within the heptahelical transmembrane domain (TMD) of the receptor (Fig. 1). Since experimental 3D structures of the TMD and/or the full-length TSHR are still missing, respective molecular models are based on available crystal structures of complexes between ectodomain fragments and hormones such FSHR/FSH (27,28) and a partial TSHR ectodomain bound with an activating or inactivating antibody (29,30). TSH and TSAb binding leads to structural alteration of the diverse extracellular parts, which finally converge at a so-called intramolecular agonist or tethered ligand (Fig. 1, dark magenta) that was previously assumed (31) and recently confirmed by peptide mapping (32) at the extracellular hinge region (magenta Fig. 1) just prior to transmembrane helix 1.
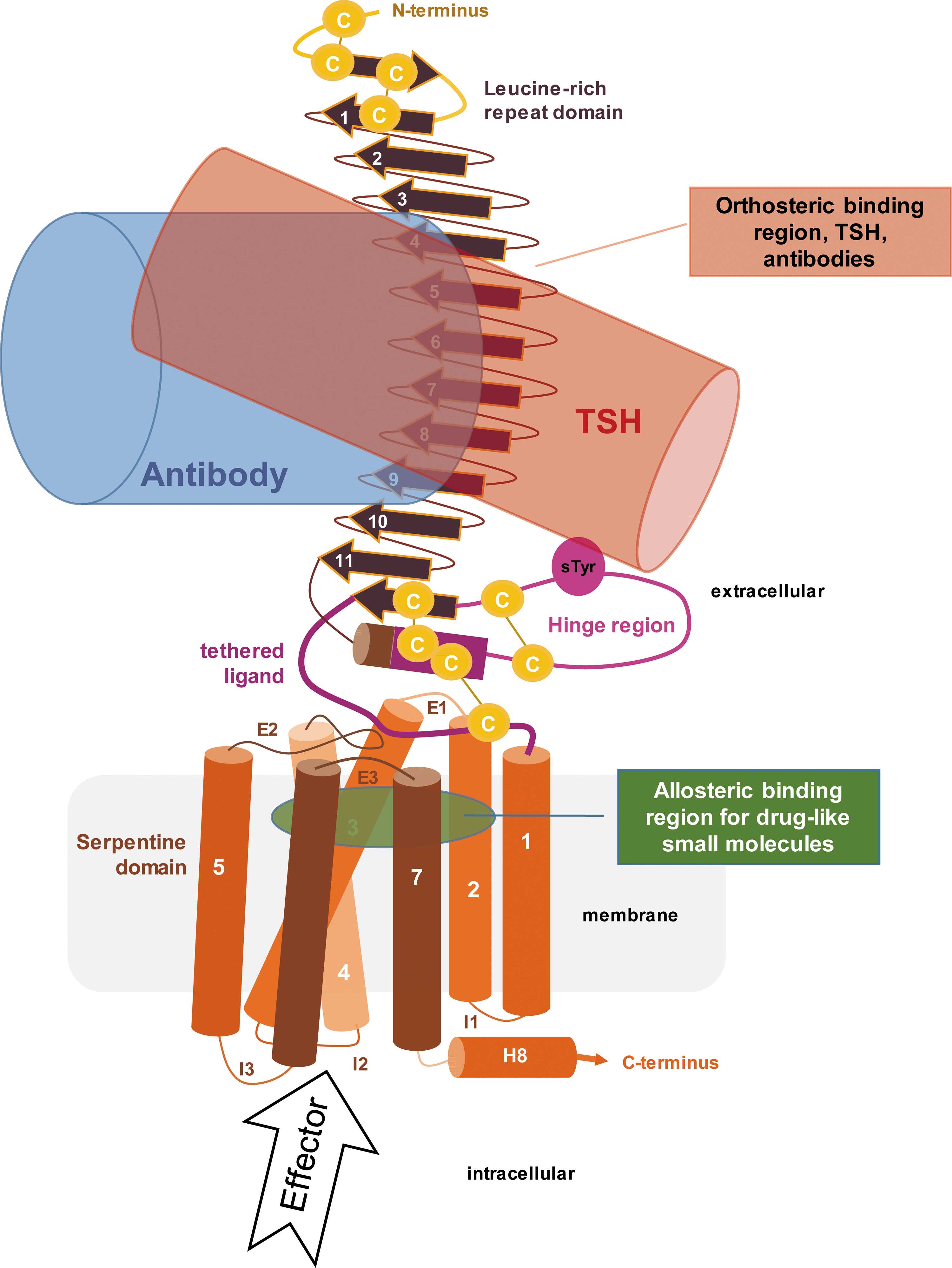
Scheme of the thyrotropin receptor (TSHR). Thyrotropin (TSH; red) and activating antibodies (blue) bind to the extracellular leucine-rich-repeat domain (brown) and hinge region (magenta). Both activate the TSHR via an internal tethered agonist (dark magenta) C-terminally located in the hinge region. Activation recruits specific effectors, leading to diverse cellular responses. Allosteric ligands bind potentially into a cleft (green) between the transmembrane helices (orange) and extracellular loops by activating (agonist) or inhibiting (antagonist) TSHR.
Druggability of the TSHR has been demonstrated previously by allosteric small-molecule ligands acting as agonists (C2) (33), or inhibiting TSHR-related hyper-stimulation (24,34,35). These include the inverse agonist Antag3 (36) and the antagonist K14 (25). Both inhibit the TSHR in micromolar concentrations. For Antag3, the inhibition of TSH and M22 (monoclonal TSHR stimulating antibody) (37) induced thyroid function was also demonstrated in vivo in mice (36). However, a mild cross-reactivity was observed for Antag3 toward the FSHR (36) and for K14 toward the LHCGR (25). For the tetrahydroquinoline compound Org274179-0, TSHR antagonism (38) and immunoglobulin G and M22 inhibition (39) with a nanomolar affinity was reported. Unfortunately, Org274179-0 also interacts with the FSHR, precluding its application for a TSHR-related disease.
Several TSHR activating and blocking antibodies are known as specific biomarkers in clinical diagnosis of GD and GO (40,41). TSHR activating antibodies titers tend to correlate with the severity of GD and GO (42 –44).
The sequence-structure-function-analysis of GPHR can be assisted by the resource
Methods
High-throughput screening
A total of 16,544 compounds of the ChemBioNet compound collection (47) were screened in 384-well format using the commercially available HitHunter™ cAMP XS assay (DiscoverX). Cells were seeded onto a 384-well plate (3707; Corning) in 40 μL of medium at a density of 15,000 cells/well. Seeded plates were then incubated overnight (37°C, 5% CO2, 95% humidity). The cell medium was removed, and cells were washed 1 × with 80 μL of phosphate-buffered saline (PBS) using a microplate washer (EL406; Biotek). Antibody working solution (20 μL; prepared by adding one part antibody solution to two parts serum-free medium/PBS) was added to each well using a dispenser (MicroFLo; Biotek). Compound (0.2 μL; 1 mM in dimethyl sulfoxide [DMSO]) was added to the plates using an automated liquid handler equipped with a 384-channel pipetting head with fixed tips (Tecan), resulting in a final compound concentration of 10 μM. Cells were incubated with compounds for 1 h at 37°C. For induction, 5 μL of bovine TSH (bTSH; Sigma–Aldrich) solution was added to the microtiter plate to columns 1–23 (final bTSH concentration 9.8 mIU/mL). Plates were incubated for another hour at 37°C. Cell lysis and detection was then carried out according to the manufacturers' protocol: 20 μL of ED/Lysis/CL working solution was added to each well of the plate, and the plate was then incubated for 1 h at room temperature. EA reagent (20 μL) was added to all wells of the plate, and the plates were incubated overnight at room temperature. Luminescence signals were detected using a plate reader (Safire2; Tecan) with 500 ms integration time and three reads per well.
The obtained data were normalized separately for each plate using robust statistics (48). Percent activity values were calculated for each well using the medians of the control wells in column 23 (bTSH-induced cells, 100% activity) and column 24 (non-induced cells, 0% activity). Z-score values indicating the distance of a sample's signal from the plate median in units of standard deviation were calculated for all sample wells of the plate. Z′-factor values for estimating the effective signal window were calculated using the control wells (49).
Concentration-dependent validation using high-throughput screening protocol
Compounds showing activity in primary screening (3 μL) were picked from the compound store and rearranged onto a 384-well plate at a concentration of 10 mM/DMSO. The compounds were then serially diluted across plates by adding 3 μL of DMSO to the plate, mixing, and transferring 3 μL onto a dry 384-well plate. Nine serial dilutions were created in total, resulting in a concentration range of 0.2–50 μM after transfer into the assay plates. The concentration-dependent assay was conducted exactly as for primary screening but with two replicates at each compound concentration. For data evaluation, the mean percent activity of the two replicates was calculated and then plotted against the compound concentration. IC50 curve fits were calculated using Pipeline Pilot's (Biovia) Least-Squares Curve Fit module fitting to a four-parameter logistic and using an IRLS algorithm.
Stereo-selective synthesis
The synthesis of the S4 diastereomers started with the opening of phthalic anhydride with aniline. Ring closure to N-phenyl-5-norborne-2,3-carboximide was accomplished in Ac2O under reflux. 4-Oxo-2-thiazolidinone was converted with Lawesson-reagent to 4-thioxo-2-thiazolidinone followed by Knoevenagel condensation with benzaldehyde in glacial acid to yield 5-ylidene-4-thioxo-2-thiazolidine. The reactive sulfur atom at the 4-position of 5-ylidene-4-thioxo-2-thiazolidine allows it to be used as heterodiene together with N-phenyl-5-norborne-2,3-carboximide as dienophile in a highly stereo-selective hetero-Diels-Alder (Fig. 2) to form the pentacyclic fused heterosystem of (4aS,5S,5aR,8aR,9R,9aS,10R and 4aR,5R,5aR,8aR,9S,9aR,10S)-7,10-diphenyl-3,4a,5,5a,8a,9,9a,10-octahydro-5,9-methano-thiazolo-(5′,4′:5,6)thiopyrano(2,3-f)isoindole-2,6,8(7H)-triones as enantiomers, which were subsequently separated (Fig. 3A and C) using chiral chromatography.

Stereo-selective synthesis route of S37.
Stereoisomers S37 (
Chiral high performance liquid chromatography
Separation of the S37 racemate (S37 rac) was performed on an analytical high performance liquid chromatography (HPLC) instrument (Agilent 1200 Series) using a Nucleocel DELTA-RP (S) column at 30°C. S37 (100 μL; ∼1 g/L) dissolved in acetonitrile/water (50/50) was injected. A clear separation was achieved by isocratic elution with acetonitrile/water (70/30) as mobile phase. The enantiomers were detected with a diode array detector at 254 nm. At a flow rate of the mobile phase of 1 mL/min, the first enantiomer eluted with a maximum at 5.1 min and was called S37a; the other eluted at 6.0 min was called S37b. Separation was performed manually from 5.0 to 5.4 min and 5.9 to 6.0 min for the first and second enantiomer, respectively. For the in vivo pharmacokinetic (PK) studies, synthesis of S37 and separation into the enantiomers on a larger scale were performed by ChemDiv. Concordance with the authors' own synthesis and separation was confirmed by HPLC and functionally by cAMP measurement in cells.
Circular dichroism (CD) spectroscopy
S37 rac and the enantiomers S37a and S37b were solved in acetonitrile at a concentration of 2 mM and were measured with the Jasco J-720 CD spectrophotometer in a 1 mm fused silica cuvette. Wavelengths from 350 to 225 nm with a 1 nm band width were applied. Data output was ellipticity in mdeg.
For the calculation of the CD spectrum for the enantiomer (4aS,5S,5aR,8aR,9R,9aS,10R)-7,10-diphenyl-5,5a,8a,9,9a,10-hexahydro-5,9-methanothiazolo(5′,4′:5,6)thiopyrano(2,3-f)isoindole-2,6,8(3H,4aH,7H)-trione, the minimized structure was optimized with density functional theory (DFT) calculations at the B3LYP/6-31G(d) level of theory. Time-dependent DFT was subsequently used with the basis set B3LYP/6-31G(d) to calculate the spin-allowed excitation energies and rotatory (Rn) and oscillator strengths (fn) of the lowest 50 excited states. All the DFT calculations were performed with Gaussian 09 software (50).
Cloning of hTSHR-GFP and mTSHR-GFP
Wild-type human TSHR (hTSHR) cDNA present in the pEGFP-N1 vector (Clontech) was cloned, as described previously (51). In the construct, green fluorescent protein (GFP) was fused C-terminally to the TSHR.
Wild-type murine TSHR (mTSHR) cDNA present in the pSVL vector was a gift from Sandra Huth and Holger Jäschke (University of Leipzig, Germany). mTSHR was amplified by polymerase chain reaction (PCR; forward primer 5′-TAT TCT AGC GCT ATG AGG CCA GGG-3′, reverse primer 5′-CTG CAG AAT TCC CAA GGC TGT TTG C-3′), thereby introducing an AfeI and an EcoRI restriction site. The PCR product was digested with the respective enzymes and cloned into the pEGFP-N1 vector digested with the same enzymes. In the resulting construct, GFP was fused C-terminally to the mTSHR. Identity of both constructs was verified by DNA sequencing (Source BioScience).
Cell culture
HEK293T cells (DSMZ) were cultivated in Dulbecco's modified Eagle's medium (DMEM) containing 1 g/L of glucose, 10% fetal bovine serum (FBS; Biochrom), 100 IU/mL of penicillin, and 100 μg/mL of streptomycin at 37°C in a humidified 5% CO2 incubator. For transient transfection of HEK293T cells, a mixture of 1 μg polyethylenimine (PEI) and 0.4 μg plasmid DNA in serum-free DMEM was added to cells grown in 24-well plates 1 day after seeding.
HEK293T cell lines stably expressing the hTSHR (HEK-TSHR) or FSHR or LHCGR have been described previously (52). The latter two cell clones were used to assess for selectivity. As an additional negative control, HEK293T cells stably expressing the construct CRF1R.Kaede (53) were used (the CRF1R is a Gs-coupled receptor unrelated to GPHRs). Stable cell lines were grown in DMEM containing 400 μg/mL of G418 and penicillin, streptomycin, and FBS as described above.
The U2OS-TSHR(L)/β-arrestin-1 stable cell line was purchased from DiscoverX and cultured with the appropriate AssayComplete Cell Culture Kit 10 according to the manufacturer's instructions.
Ligand treatment and determination of intracellular cAMP accumulation
On a 24-well plate, 2 × 105 cells/well were seeded without selection antibiotics. Seventy-two hours after seeding and 48 hours after transfection, cells were treated with ligands and intracellular cAMP accumulation was measured by radioimmunoassay, as described previously (54). Shortly after, cells were washed with 1 mL of stimulation buffer (DMEM supplemented with 10 mM of HEPES, 0.5% BSA, and 0.25 mM of 3-isobutyl-1-methylxanthine) and incubated for 1 h at 37°C with stimulation buffer alone or stimulation buffer containing hormones (bTSH, recombinant human FSH [R&D systems], human chorionic gonadotropin [Merck]), antibodies (M22 [RSR Ltd.], KSAb1 [gift from Paul Banga, University of Essen, Germany]), and/or small-molecule ligands at indicated concentrations. Small-molecule TSHR ligands C2 and Antag3 were a gift from Marvin Gershengorn (NIH, Bethesda, MA). N′-(4-[2-phenyl-1H-indol-3-yl]-1,3-thiazol-2-yl)guanidine referring to the antagonist K14 depicted in Latif et al. (25) was purchased from Enamine. For ex vivo experiments, HEK-TSHR cells were treated with serum from GO patients at a 1:10 dilution in stimulation buffer for 1 h at 37°C. Serum samples were collected from GD patients with various stages of clinically apparent GO referred to the Department of Ophthalmology at the University Hospital Essen. The diagnosis of GO as well as the thyrotropin receptor antibody (TRAb) measurements have been described previously (55,56). Serum samples from patients with mild and moderate-severe GO and from healthy controls were used. Serum samples of GO patients showed TRAb values between 14 and >40 IU/mL, whereas serum from controls was negative for TRAbs. The investigations were conducted in accordance with the Declaration of Helsinki, and informed consent was obtained from the patients. Study procedures were formally approved by the ethical review committee of the University of Essen.
Determination of β-arrestin-1 recruitment to TSHR
β-Arrestin-1 recruitment to TSHR after activation with bTSH or additional inhibition with S37a was determined in the U2OS-TSHR(L)/β-arrestin-1 cell line with the PathHunter enzyme fragment complementation assay (DiscoverX). Twenty-four hours before the experiment, 2.0 × 104 cells/well were seeded onto a 96-well plate in AssayComplete Cell Plating 5 reagent. bTSH (1 × 10–4 to 0.5 IU/mL) and S37a (1–50 μM for each bTSH concentration–response curve) or DMSO were diluted in Cell Plating 5 reagent and added to the cells. After 90 min of incubation at 37°C in a 5% CO2 humidified atmosphere, β-arrestin-1 recruitment was quantified using the PathHunter Detection Kit according to the manufacturer's instructions. Luminescence was measured by the luminescence plate reader Tecan Infinite F200 Pro.
alamarBlue cell viability assay
HEK293T-TSHR cells (8 × 104 per well) were seeded onto a 96-well plate and grown for 48 h. The cells were treated for 18 h with S37 ranging from 10 to 100 μM or with the equivalent concentration of DMSO as solvent control. Cell viability was measured using the alamarBlue assay (Invitrogen) according to the manufacturer's instructions.
PK studies in mice
For PK studies, the contract research company Pharmacelsus GmbH was commissioned. All experimental procedures were approved by and conducted in accordance with the regulations of the local animal welfare authorities (Landesamt für Gesundheit und Verbraucherschutz, Abteilung Lebensmittel- und Veterinärwesen). SWISS (CD1) mice (Janvier Labs) were housed in a temperature-controlled room (20–24°C) and maintained on a 12 h/12 h light/dark cycle. Food and water were provided ad libitum.
In an initial PK study, mice (body weight 38–43 g) were treated with a single application of S37a by either intravenous (i.v.) injection (1 mg/kg in DMSO/PEG400; 20:80) into the lateral tail vein or oral (p.o.) application by gavage (10 mg/kg in 1% Tween 80/methylcellulose 0.5%). At six time points after application, the blood of three mice per time point was sampled as lithium heparin whole blood from retrobulbar venous plexus.
In a follow-up PK study, mice (body weight 33–39 g) were treated by a gavage of 50, 100, or 150 mg/kg S37a in 1% Tween 80/methylcellulose 0.5%. The blood of three animals per dosing group was sampled 15 min and 7 h after application. S37a content in plasma was quantified by liquid chromatography–mass spectrometry in each study.
Results
High-throughput screening and stereo-selective synthesis resulting in the new SMA S37a for TSHR
This study aimed to identify new molecular scaffolds of small molecules that block TSHR activation but not that of other GPHRs.
High-throughput screening of 16,544 compounds of the ChemBioNet library (47) at 10 μM compound concentration and one measurement per compound delivered 350 samples that showed a significant reduction in TSH induction, with a Z-score of <−3. The Z′-factor metrics across all plates showed a median value of 0.7, indicating excellent signal-to-noise separation of the assay. The 350 compounds were picked and reinvestigated using the same assay in the concentration range 0.2–50 μM. In order to exclude TSHR-independent inhibitors, a counter screen was conducted in TSHR-free cells stimulated by forskolin. Twelve compounds were then selected for further investigation.
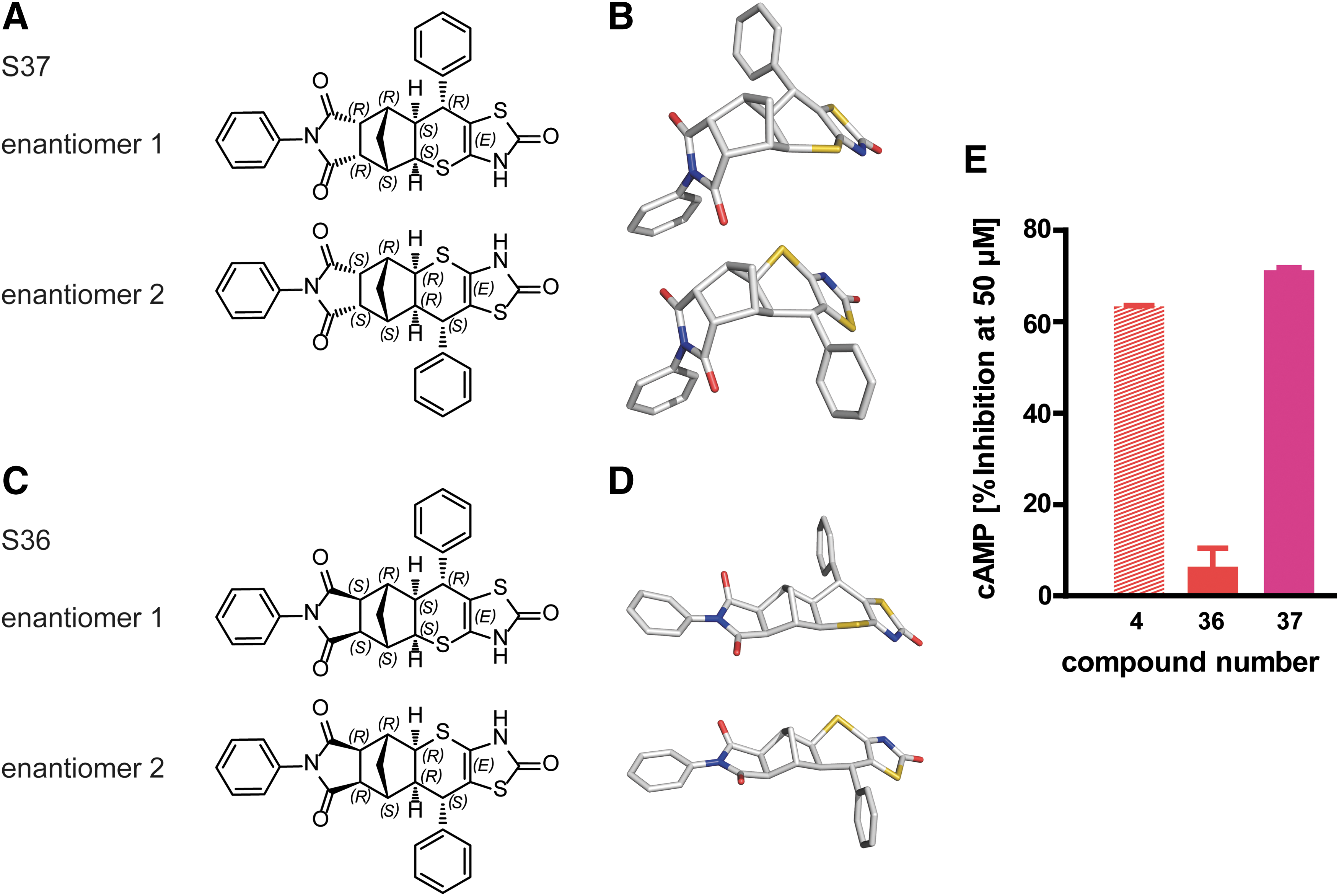
A secondary screen in HEK-TSHR cells confirmed three scaffolds that showed >50% inhibition of bTSH-induced cAMP accumulation in concentrations up to 50 μM. Here, the focus was on the hit S4 with unknown stereo-selectivity for further subsequent structure–activity relationship studies. S4 contains seven chiral centers. Therefore, a stereo-selective synthesis of the S4 was developed to produce diastereomers S36 and S37 (Fig. 3), starting from prochiral exo or endo phthalic anhydride. Indeed, these chiral selective compounds S36 and S37 exhibit a clear separation of the functional effect. The endo-stereoisomer S37 (Fig. 3A and B) has an antagonistic effect of 70% at 50 μM (Fig. 3E), whereas the second isomer, S36, which is in exo-conformation (Fig. 3C and D), does not have an antagonistic effect at all. Figure 3 highlights the conformational difference of the two stereoisomers. S36 exhibits an extended conformation, whereas S37 adopts a bent conformation that is probably responsible for its highly selective function.
Enantiomeric separation of S37 enhanced affinity of enantiopure molecule S37a
The enantiomeric separation of the still racemic compound S37 into enantiomers S37a and S37b revealed that S37a has 20% stronger inhibition than S37, while S37b had only a minor effect (Fig. 4A). By comparing experimental electronic circular dichroism spectra (Fig. 4B) and the predicted one using time-dependent DFT calculation at the B3LYP/6-31G(d) level of theory, the absolute configuration was assigned to the stronger inhibiting enantiopure structure of S37a (Fig. 4C).
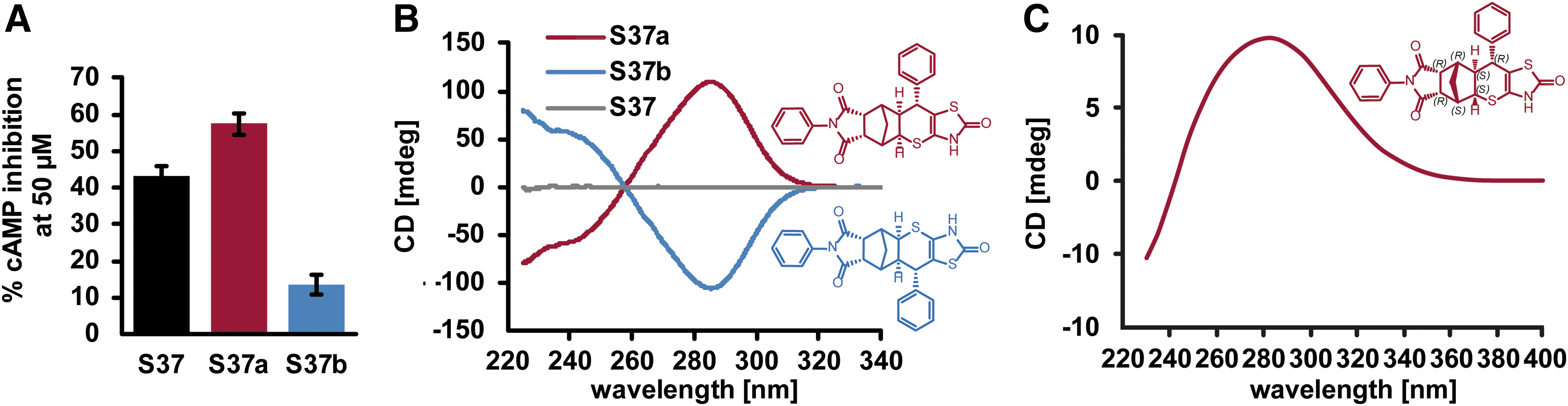
Assignment of two enantiomers S37a and S37b to their absolute configurations. (
Effect on other signaling pathways
Hormone (TSH) induced TSHR activation revealed different inhibition patterns by S37 or S37a for cAMP production (Gs activation) and β-arrestin-1 recruitment, respectively. The bTSH concentration–response curve for cAMP was right-shifted by increasing S37 concentrations (Fig. 5A), suggesting a competitive antagonism of S37 and TSH. In the case of β-arrestin-1 recruitment, however, the maximal effect was reduced by increasing S37a concentrations (Fig. 5B). The latter result indicates a noncompetitive antagonism of S37 and TSH in an allosteric mechanism. These results only seem to be contradictory at first sight. In fact, they strongly suggest that TSH induces Gs and β-arrestin-1 coupling by spatially distinct intramolecular routes in TSHR.
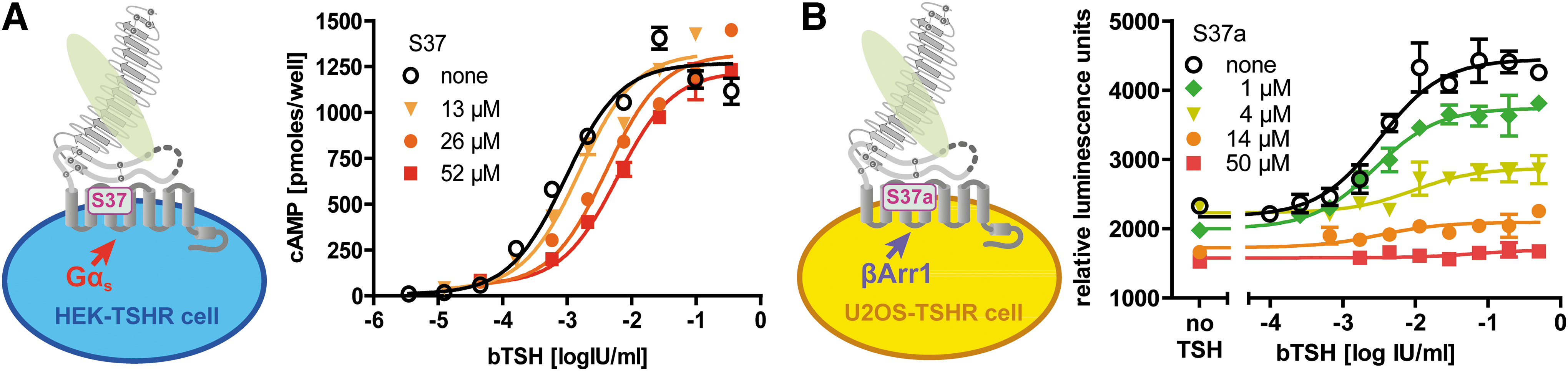
Concentration-dependent inhibition of TSH-induced TSHR activation by S37 in competition experiments. (
S37a is highly selective for TSHR
The antagonistic activity of compounds S37a and, as reference compounds, the small-molecule inhibitor K14 depicted in (25) and the micromolar inverse agonist Antag3 of the NIH (36) were tested on the homologous members of the GPHR family, the FSHR and LHCGR (Fig. 6). As a negative control, the CRF1R, a Gs-coupled family B GPCR, was tested as well. All receptors were stably expressed in HEK-293T cells, and the impact of each substance was measured upon administration of 50 μM of the respective hormone alone or together with the compound. For comparison, antagonistic potential of each compound was calculated. In the case of the negative control (CRF1R), no antagonistic effects of the compounds were observed. The antagonistic effect on cAMP formation of the new compound S37a was highly TSHR selective, even in the micromolar concentration range used in these experiments. The reference substance Antag3 also exhibits a TSHR preference. However, it affects the FSHR in micromolar concentrations too. The second reference substance referring to K14 (25) is a strong but not highly TSHR-selective inhibitor.
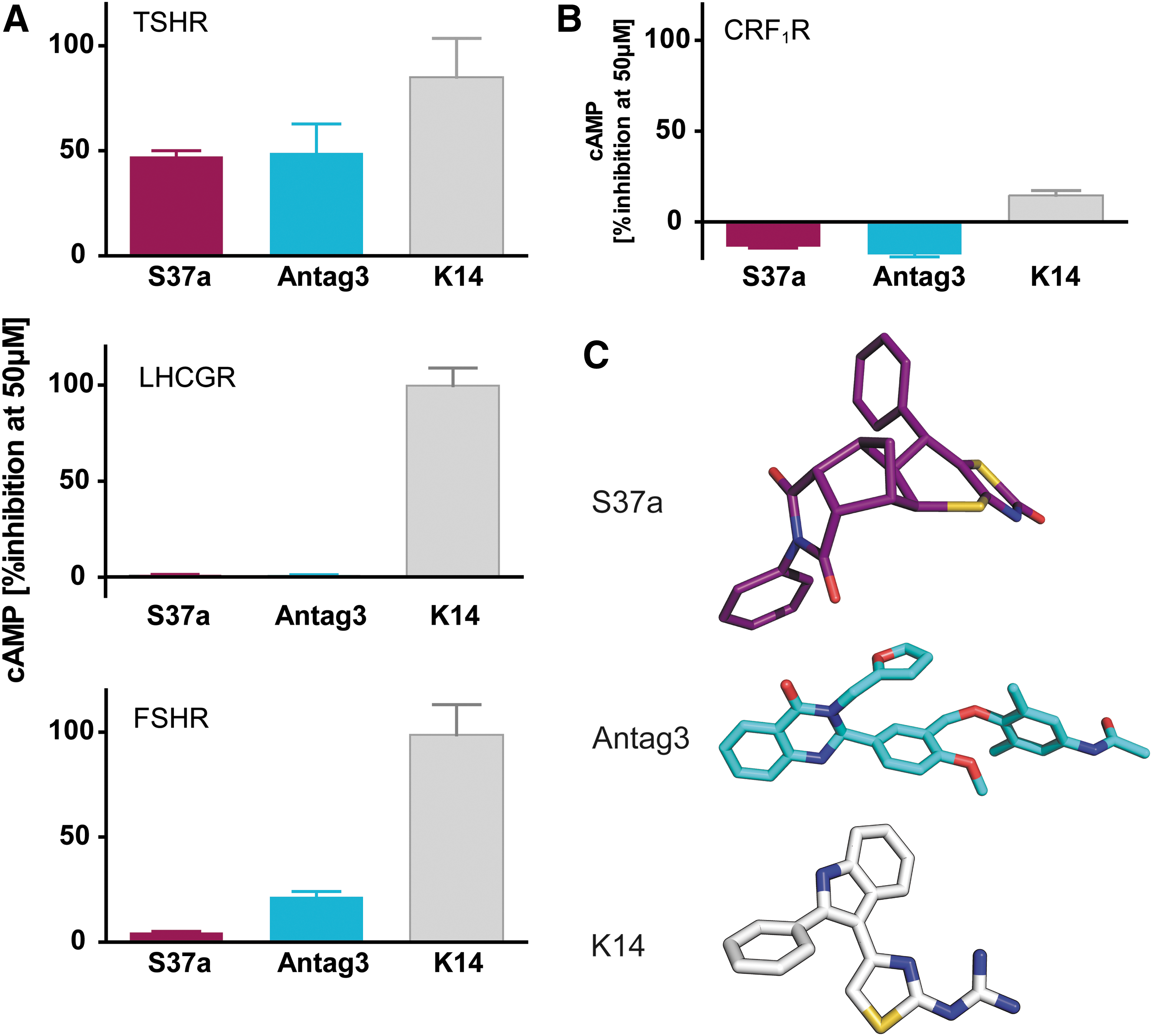
Antagonistic effect (% cAMP inhibition) on TSHR and homologous receptors for lutropin and follitropin. (
Inhibition of stimulating monoclonal antibodies and small-molecule agonist
Next, the study tested whether the new compound S37a is also able to block antibody-mediated TSHR activation. S37a inhibits cAMP activation by the monoclonal TSHR-activating antibody M22 or TSH with similar potency (Fig. 7). The substance could also block the activation by the mouse monoclonal antibody KSAb1 (gift from Paul Banga) (57) (Fig. 7A). Of note, S37a has only barely detectable agonistic properties (Fig. 7A).

Effect of S37a on human and mouse thyroid stimulating antibodies (TSAb) and small-molecule agonist C2. (
S37 also reduces cAMP activation induced by the small-molecule agonist C2 (Fig. 7B), which has been reported to bind into an allosteric binding pocket in the TMD (33). Albeit C2 was inhibited using the racemate in transiently transfected HEK-TSHR-GFP cells, the inhibitory pattern is comparable to that shown for TSH and the antibodies.
S37a also blocks TSHR, which was stimulated by the sera of GO patients
Under pathogenic conditions, it is critical to inhibit the diverse, oligoclonal TSHR activating antibodies that are responsible for the TSHR dysregulation in GO (58). In order to evaluate the effect of S37a in a disease-related scenario, next the study tested the inhibitory effect of S37a on the stimulation of the TSHR mediated by TSAb-containing sera of patients suffering from GO.
Ex vivo experiments in HEK-TSHR cells revealed that cAMP is activated differently by the individual sera (according to TRAb values, not shown) up to an activation that is comparable to TSH (blue, Fig. 8). Most importantly, the study shows that all sera with activated cAMP were inhibited up to 50–60% by S37a (red Fig. 8), suggesting a clinically applicable mode of action. The results verified TSHR as a therapeutic target for treating GO using the inhibitor S37a.
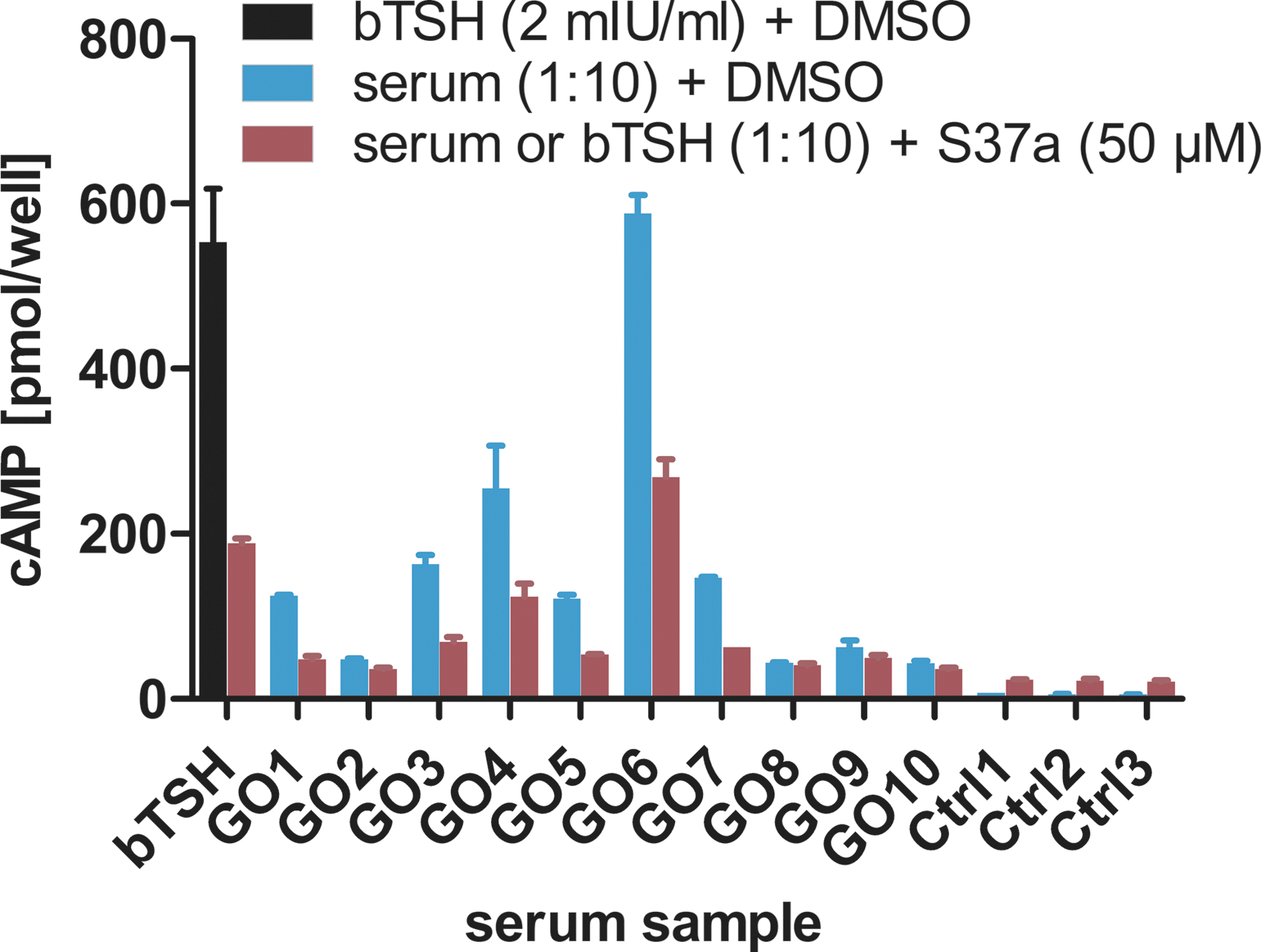
S37a inhibits TSHR-cAMP-activating sera derived from Graves' orbitopathy (GO) patients. Stable HEK-TSHR cells were treated with TSHR-activating sera (GO patients) in a 10-fold dilution or bTSH (2 mIU/mL) as a positive control. As a negative control (Ctrl), sera from healthy patients were used. With the addition of 50 μM of S37a, cAMP accumulation induced by GO sera was inhibited by 50–60% compared to bTSH-treated cells.
In vitro and in vivo tolerance and applicability
S37 does not impair cell viability in vitro
The racemate S37 was tested for its toxicity by the alamarBlue test using the Hek293T-TSHR cell line. The concentration-dependent (10–100 μM) toxicity test did not reveal any toxic effect for the cells upon treatment with S37 (data not shown). Therefore, it was concluded that substance S37 does not affect cell viability in the concentration range evaluated by this assay.
The inhibitory effects of S37a on mTSHR and hTSHR are comparable
Prior to in vivo mouse experiments, the intention was to confirm that the potency of S37a on mTSHR and hTSHR activation is comparable. GFP-tagged mTSHR was transiently expressed in HEK293 cells. Expression and plasma membrane localization was confirmed by confocal LSM (data not shown). mTSHR could be stimulated by bTSH in a concentration-dependent manner with a slightly higher EC50 than hTSHR (1.3 and 0.8 mIU/mL, respectively; Fig. 9). mTSHR was inhibited by S37a with an IC50 of 40 μM; hTSHR S37a showed an IC50 of approximately 20 μM.
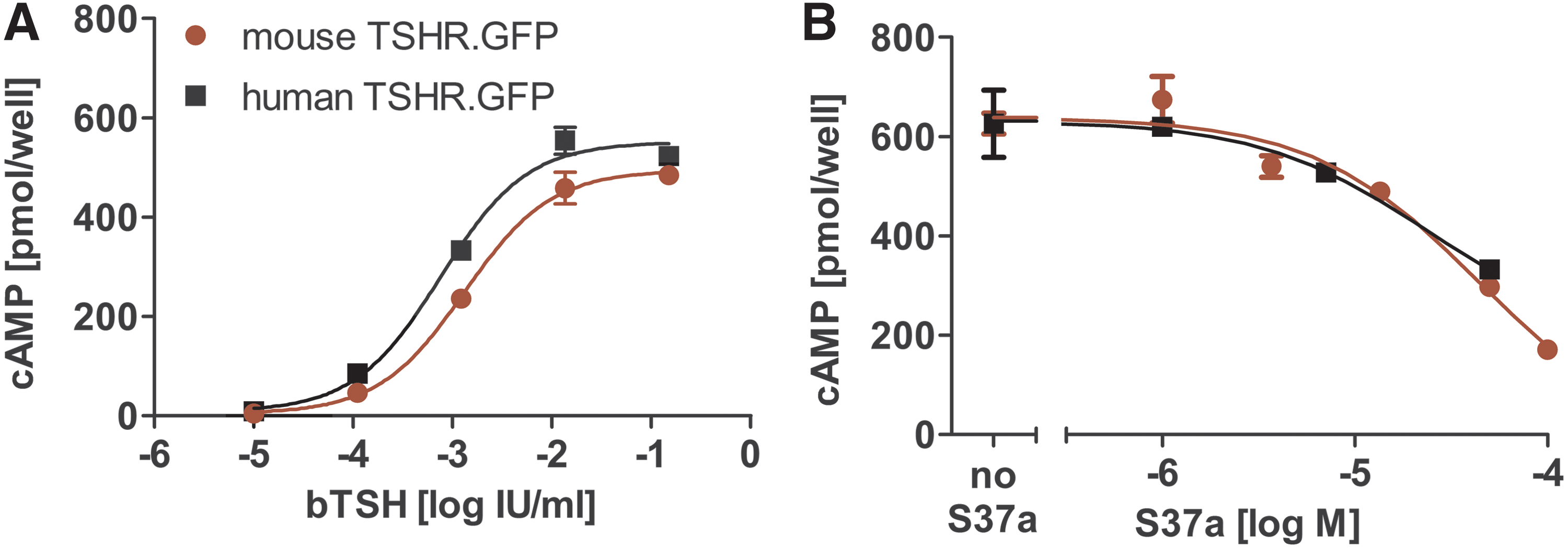
Comparison of mTSHR and hTSHR signaling. (
S37a PK in mice
In an initial in vivo PK and tolerance study, a single low dose was administered to mice i.v. (2.5 mg/kg) and p.o. (10 mg/kg). The S37a plasma concentration time curves (Fig. 10A) show a remarkable 53% oral bioavailability, as well as a half-life of 2.9 h after oral application. In a second step, higher oral doses (50, 100, and 150 mg/kg) were applied. Plasma concentrations up to 6000 ng/mL, corresponding to 13 μM of S37a, were reached 7 h after application of 150 mg/kg. A dose-dependent increase of S37a plasma concentrations from 0.25 and 7.0 h after oral administration was observed (Fig. 10B). The compound was also well tolerated at the highest dosage, and no clinical adverse effects were observed.

S37a in vivo pharmacokinetics in mice. S37a plasma concentration over time for (
Discussion
A potential approach to bridge the described therapeutic gap in GO are SMA targeting proteins involved in GO pathogenesis. The TSHR is reckoned to be a potential target for pharmacological intervention of GD and GO (14,59 –62). An inflammatory process (63) and IGF-1 signaling are also thought to be involved in the pathogenesis of GO (59 –62,64,65). Until now, no specific treatment has been available for patients with GO that has the potential to prevent manifestations such as proptosis due to adipogenesis and/or impaired eye muscle function due to fibrosis (8). In vivo and in vitro experiments have revealed that TSHR stimulation (7) together with TSHR and IGF1-R cross-talk (61,62,66) play an important role in the pathogenesis of GO and that the development of a targeted therapy for severe cases of GO appears to become realistic.
In a Phase II study with the IGF1R blocker Teprotumumab, a remarkable effect was shown, especially in terms of proptosis and inflammation reduction (67). However, complete remission did not occur in all patients. The question remains whether direct blocking of TSAb action at the TSHR is more efficient for GO treatment. Such a mechanism of action would of course also represent a most welcome alternative for the treatment of hyperthyroidism.
Using high-throughput screening and relative stereochemistry, a new small molecule with challenging synthesis due to seven chiral centers as an inhibitor and negative allosteric modulator of TSHR has been identified as a potential new drug for the treatment of Graves' hyperthyroidism and orbitopathy, namely S37a. The bent-shaped molecule stereoisomer S37 exhibits in contrast to the straight-shaped inactive isomer S36 (Fig. 3) a micro-molar inhibition of TSH-induced cAMP accumulation in HEK and Chinese hamster ovary cells expressing hTSHR.
Concerning the different pharmacological patterns of S37a signaling (inhibiting TSH competitively in cAMP and noncompetitively in β-arrestin-1 assay), two explanations may be discussed. First, spatially distinct intramolecular routes may be present for Gs and β-arrestin-1 activation in the TSHR (Fig. 11). Since small-molecule agonists have been shown to act allosterically within the TMD (33), the observed competition of S37 with TSH in the cAMP assay (Fig. 5A) was rather unexpected. However, activation by TSH may be conveyed in this case by an intramolecular tethered agonist (32) prior to TMD 1 (Fig. 1), which is assumed to be located between the extracellular loops (31) close to the allosteric binding pocket in the TMD. S37a may block conveying the signal for cAMP activation by the internal tethered agonistic unit. A second possible explanation for the observed competitive and noncompetitive antagonism could be a different course of intracellular persistent signaling for cAMP (68,69) and β-arrestin-1, as S37a may also bind to intracellular receptors.

Scheme of how TSH may activate TSHR by spatially distinct intramolecular routes for Gs and β-arrestin-1. Route 1 (red dashed) mediates Gs coupling. S37a could hamper this mechanism via internal tethered agonist. Route II (blue dashed) mediates β-arrestin-1 recruitment, which can be inhibited by S37a as negative allosteric modulator.
The results allow focused suggestions to be made for further stereo-selective synthesis to optimize the antagonistic effects. Enantiomeric separation of the still racemic molecule S37 resulted in enhanced affinity for the enantiopure molecule S37a (Fig. 4).
The IC50 value of S37a is in the low micromolar range, and thus it is comparably potent in inhibiting TSHR as the two reference compounds Antag3 (36) and K14 (25). However, previously identified discriminative pharmacophores covering the allosteric binding pocket of the TSHR (70) based on molecular sampling of signaling-sensitive (54) and -silencing (71) residues might support further optimization of S37a in the future.
Of note, S37a is the first structure with antagonistic properties, which is exclusively highly TSHR selective, since it does not affect the other GPHRs, namely the FSHR and LHCGR. This advantage in comparison to the two reference compounds (Fig. 6) is probably due to the sterically challenging, rigid, and bent shape of the enantiopure molecule S37a, whereas the others with different scaffolds are either flat or more flexible (Fig. 6C). In the assay with stably transfected HEK293 cells of the three GPHR, K14 showed a stronger effect on FSHR and LHCGR than that found by Latif et al. (25). A reason could be that different cell lines expressing the three GPHRs were used.
This study has excluded the interaction of S37a with the CRF1 receptor, which is also a Gs-coupled receptor but belongs to class B GPCR and is thus unrelated to GPHRs. At the present time, side effects on other proteins cannot be totally excluded. Samples of a wide range of different GPCRs have to be considered in further studies. However, the fact that there are many drugs on the market acting specifically on one GPCR implies that evolution has fine-tuned the GPCR to an extent that they can be targeted specifically. Structure and sequence of the TMD of GPHR was obviously only under a milder evolutionary pressure, since the different hormones bind extracellularly. The pronounced sequence homology of the TMD among GPHR might explain why, for an allosteric small ligand, a high specificity toward a particular GPHR is hard to achieve. Thus, clarification of the detailed binding site of S37a in the future can help to identify the precise structural features of the TSHR that are crucial for the high selectivity of S37a.
S37a is a negative allosteric modulator in in vitro assays for (i) the hormone TSH and (ii) monoclonal antibodies M22 (human) and KSAb1 (murine), as well as for (iii) small-molecule allosteric agonist C2. Moreover, ex vivo studies using GO patients' sera in HEK cells expressing TSHR show that S37a also inhibits cAMP activation by diverse pathogenic TSAb (Fig. 8). TSHR antibodies in patients with Graves' hyperthyroidism are probably oligoclonal (58). Therefore, from a structural point of view, it is important that SMA bind downstream of the antibodies or the hormone binding site, as they probably would not cover the paratopes of all different antibodies. From a therapeutic point of view, it is noteworthy that stronger cAMP inhibition by S37a is observed in sera with higher TSAb values of patients with severe GO (sera 3, 4, 6, and 7 in Fig. 8) compared to those with mild GO. Thus, the S37a effect shows a clear correlation with the severity level of GO.
Apart of its selective antagonism, S37a exhibits a slight partial agonistic effect, which has the advantage that the TSHR activity is not completely abolished, and a low cAMP level similar to that of endogenous basal receptor activity is retained.
No toxicity of S37a was observed in in vitro studies at up to 100 μM in HEK293 cells. PK in vivo mouse studies showed tolerance and 53% bioavailability and a half-life of 2.9 hours for a low dose. There is a dose-dependent increase in plasma levels of S37a after oral administration and also a longer retention time (Fig. 10).
In summary, a new highly TSHR-selective small-molecule inhibitor is presented, which is promising to develop further for targeted treatment of Graves hyperthyroidism and orbitopathy. The next steps toward this development are further optimization of potency, as well as in vitro (orbital fibroblast cell cultures) and in vivo applications.
Footnotes
Acknowledgments
We appreciate the considerable support from Heike Nikolenko with the CD spectrophotometer measurements. The small-molecule agonist C2 and inverse agonist Antag3 were provided by Susanne Neumann and Marvin Gershengorn (NIH). We thank Paul Banga (University of Essen, Germany) for the mouse monoclonal antibody KSAb1, as well as Sandra Huth and Holger Jäschke (University of Leipzig, Germany) for the mouse TSHR cDNA. This work was supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation; grant no. KR1273/4-2).
Author Disclosure Statement
P.M., I.H., E.S., J.F., M.Ne., M.Na., J.v.K., and G.K. filed a patent for the TSHR antagonist. C.R., S.S., H.S., R.S., U.B.P., and A.E. do not disclose any commercial association that might create a conflict of interest in connection with the manuscript.