
Abstract
Thyroid hormones have long been known to have a range of effects on the cardiovascular system. However, significant knowledge gaps exist concerning the precise molecular and biochemical mechanisms governing these effects and the optimal strategies for management of abnormalities in thyroid function in patients with and without preexisting cardiovascular disease. In September 2017, The National Heart, Lung, and Blood Institute convened a Working Group with the goal of developing priorities for future scientific research relating thyroid dysfunction to the progression of cardiovascular disease. The Working Group reviewed and discussed the roles of normal thyroid physiology, the consequences of thyroid dysfunction, and the effects of therapy in three cardiovascular areas: cardiac electrophysiology and arrhythmias, the vasculature and atherosclerosis, and the myocardium and heart failure. This report describes the current state of the field, outlines barriers and challenges to progress, and proposes research opportunities to advance the field, including strategies for leveraging novel approaches using omics and big data. The Working Group recommended research in three broad areas: 1) investigation into the fundamental biology relating thyroid dysfunction to the development of cardiovascular disease and into the identification of novel biomarkers of thyroid hormone action in cardiovascular tissues; 2) studies that define subgroups of patients with thyroid dysfunction amenable to specific preventive strategies and interventional therapies related to cardiovascular disease; and 3) clinical trials focused on improvement in cardiovascular performance and cardiovascular outcomes through treatment with thyroid hormone or thyromimetic drugs.
Introduction
The effects of thyroid dysfunction on the cardiovascular system have been well documented for more than two centuries (1). Clinically, both thyroid hormone excess and deficiency can induce or exacerbate cardiovascular disorders, including atrial and ventricular arrhythmias, atherosclerotic vascular disease, dyslipidemia, and heart failure, thereby contributing to higher risk of premature morbidity and death. Moreover, a growing body of observational data suggests that cardiovascular risk may also be increased in subgroups of patients with subclinical thyrotoxicosis or subclinical hypothyroidism. Heightened risk for both incident heart failure and thyroid disease in the aging population threatens a growing burden of both diseases in the coming decades, underscoring the need for greater attention to their intersection.
On the basis of a current literature review and a review of grant funding in these fields, the National Heart, Lung, and Blood Institute (NHLBI) convened a Working Group in Bethesda, MD, on September 8, 2017, to discuss current knowledge and future directions for research in thyroid status and cardiovascular disease (CVD), to stimulate research in this area, and to foster collaboration across disciplines. The Working Group was comprised of a multidisciplinary group of scientists with expertise in basic, clinical, and population sciences pertinent to thyroid and cardiovascular function and dysfunction. It reviewed and discussed the role of normal thyroid physiology, the consequences of thyroid dysfunction, and the effects of therapy in 3 cardiovascular areas: cardiac electrophysiology and arrhythmias, the vasculature and atherosclerosis, and the myocardium and heart failure. In the context of limited available data to address the intersection between thyroid dysfunction and CVD, a particular focus included the opportunities to leverage analysis of extremely large data sets (big data) to provide new insights into diagnosis, risk stratification, and management of thyroid abnormalities. Before the meeting, Working Group members participated in conference calls (2 conference calls for each of 4 subgroups) and completed worksheets structured according to the framework of this article: current state of the field, barriers and challenges, and research opportunities. From these worksheets and discussions from the Working Group meeting, National Heart, Lung, and Blood Institute program staff and the Working Group cochairs developed the recommendations outlined here, with additional refinement by the Working Group during article drafting and critical revision. Because recent review articles provide a comprehensive review of the literature on thyroid function and CVD (2,3) and guidelines provide recommendations for clinical care (Table 1), this report briefly summarizes the current state of fundamental, translation, and population science, outlines barriers and challenges to progress, and presents research opportunities to advance the field.
Guidelines with Recommendations for Management of Thyroid Dysfunction Coexistent with Cardiovascular Disease
ASCVD, atherosclerotic cardiovascular disease; CVD, cardiovascular disease; TSH, thyroid-stimulating hormone.
Thyroid status is determined through measurement of thyroid function tests in peripheral blood. Concentrations of thyroid stimulating hormone (TSH), which is produced by the pituitary gland, and the thyroid hormones thyroxine (T4) and triiodothyronine (T3), are easily measured through established assays. Figure 1 provides the classification of thyroid status using these assays. The term “subclinical hyperthyroidism” refers to abnormally low TSH concentrations with free T4 and total or free T3 concentrations within the reference range. However, observational studies frequently determine subclinical hyperthyroidism from only TSH and free T4 levels, as T3 levels are usually within the reference range if free T4 levels are. The form of overt hyperthyroidism called T3 toxicosis, in which free T4 levels are normal and T3 levels are elevated, is uncommon. “Subclinical hypothyroidism” refers to abnormally high TSH concentrations with free T4 levels within the reference range. The low T3 syndrome refers to isolated low T3 levels with free T4 and TSH levels within the reference range (often defined as 0.45–4.5 mIU/L). In addition, throughout this article, the term “hyperthyroidism” denotes thyrotoxicosis caused by both intrinsic (“endogenous”) thyroid dysfunction and excessive (“exogenous”) thyroid hormone supplementation.
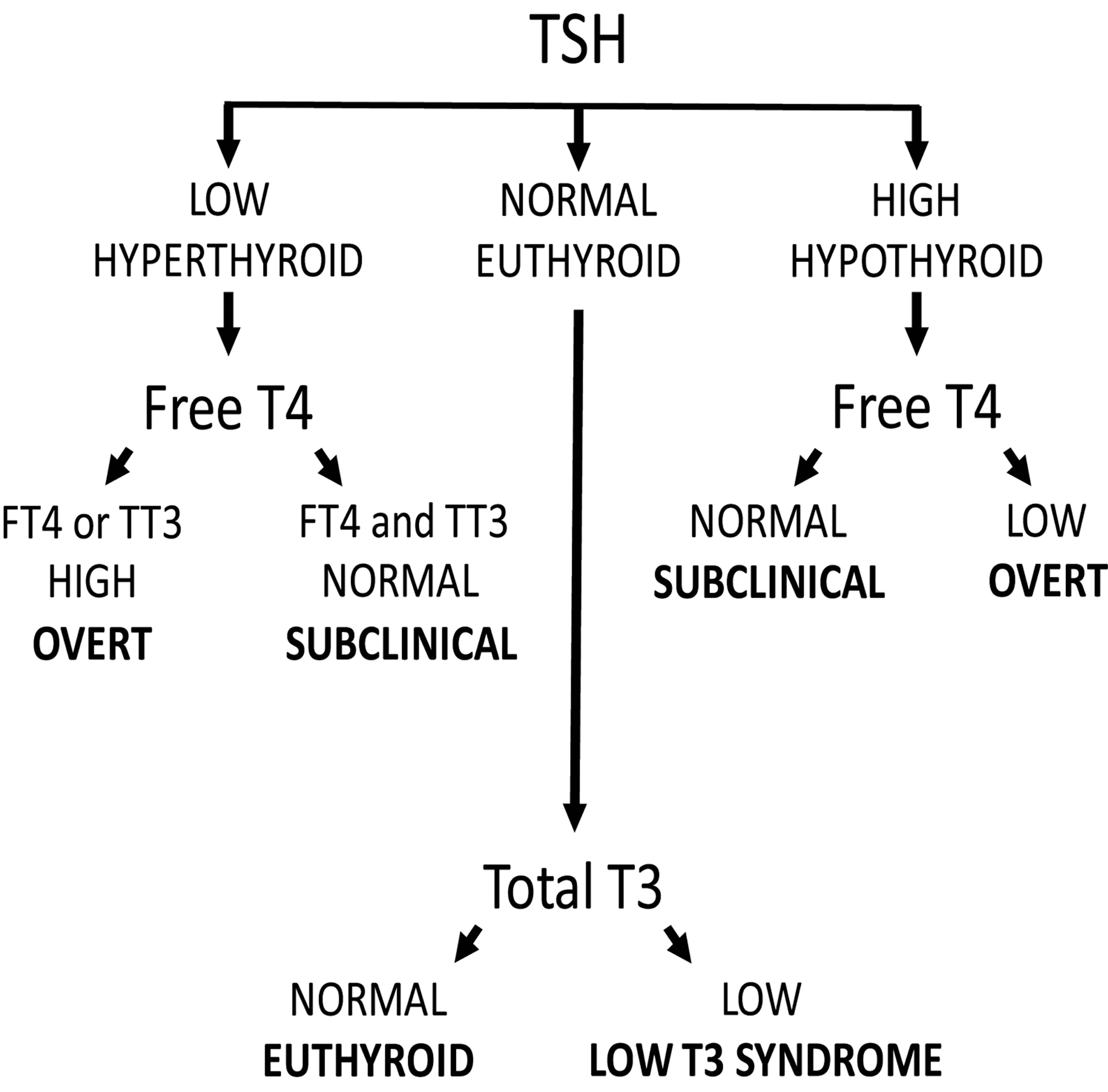
Commonly defined categories of thyroid status. FT4, free T4; TSH, thyroid-stimulating hormone; TT3, total T3.
These definitions of thyroid dysfunction are based on the population distribution of serum TSH and free T4 concentrations: levels are deemed abnormal when they are below or above the 2.5th and 97.5th percentiles, respectively. Clinically, a definition that is based on the risk of cardiovascular complications, hypothyroid or hyperthyroid complaints, and the prevention of these complications could direct treatment of thyroid disease. These clinical decision limits, or thresholds, have been used for the definition of many other cardiovascular risk factors, including obesity, dyslipidemia, hypertension, and diabetes mellitus, but they do not exist for thyroid function (14).
OMICS AND BIG DATA
In this section, we describe the role of large data consortia in refining the application of thyroid function testing to clinical decisions, followed by discussion of barriers and opportunities and research opportunities using omics and big data.
Current state of the field
The availability of extremely large data sets (big data) that can be analyzed to reveal associations, patterns, and trends in patient- or population-based cohorts provides unique opportunities in medicine. In addition, over the last decade, important progress has been made in epigenomics, transcriptomics, proteomics, and metabolomics (15). These techniques are also likely to play a key role in addressing many of the knowledge gaps in the field of thyroid dysfunction and CVD. Currently, there are two large consortia in the thyroid field: the Thyroid Studies Collaboration (16) and the ThyroidOmics Consortium (17). The Thyroid Studies Collaboration includes >75,000 participants from 20 cohorts with thyroid function measurements at baseline and prospective follow-up of cardiovascular outcomes (18
–23), and the ThyroidOmics Consortium includes >80,000 participants from 34 cohorts with available data on thyroid function and at least one of the aforementioned omics (
The personalized treatment approach, which includes information from the patient's genetic background, would aid in prescribing preventive care for CVD. In healthy individuals, serum thyroid function tests show substantial interindividual variability, leading to wide reference ranges. However, the intraindividual variability lies within a much narrower range. This indicates that every individual has a distinct hypothalamic–pituitary–thyroid (HPT) axis set point within these wide reference ranges (27). On the basis of this construct, patients could therefore still be relatively hypothyroid or hyperthyroid when serum TSH and free T4 levels are normalized to within the reference ranges but deviate from their HPT axis set point. This hypothesis is supported by various studies showing that, despite normalized TSH and free T4 levels, ∼15% of patients treated for hypothyroidism or hyperthyroidism still have significant thyroid-associated complaints (28 –32). To what extent a deviation from the HPT axis set point is also important in determining a patient's cardiovascular risk needs to be clarified in future studies. Unfortunately, it is currently not possible to predict an individual's HPT axis set point. However, twin studies have estimated that ∼60% of the variation in serum thyroid parameters is determined by genetic factors (33). In the last decade, genome-wide association studies (GWAS) have successfully identified many new genetic variants that influence TSH and free T4 levels, and the first whole-genome sequencing projects have also identified a few new relevant loci (34,35). However, these known variants together only explain 5–6% of the variation in serum TSH and free T4 levels, which means that many variants could still be awaiting discovery. Therefore, larger genome-wide association studies and whole-genome and -exome sequencing studies are needed to identify the remainder of this missing heritability. The identification of these variants is not only important for HPT axis set-point prediction, but also key to performing reliable Mendelian randomization studies (Fig. 2). Mendelian randomization is a statistical approach using genetic variants to test whether an intermediate phenotype (e.g., thyroid function) is causally related to another phenotype (e.g., coronary heart disease; CHD) (36). This method has the potential to prove direct causality. These studies are particularly needed in the field of thyroid function and cardiovascular complications. Thyroid dysfunction has already been associated with various complications in epidemiological studies, but for many disorders, including CHD and heart failure, direct causality remains unproven. A recent Mendelian randomization study on thyroid function and ischemic heart disease found no effects with genetic determinants of free T4, TSH, or anti-thyroid peroxidase antibodies (37). Despite the large sample size, this study was limited by the small number of genetic variants used. Therefore, Mendelian randomization studies performed in large consortia using more genetic markers are needed to provide clear insights into the true effects of thyroid function on the risk of cardiovascular complications.
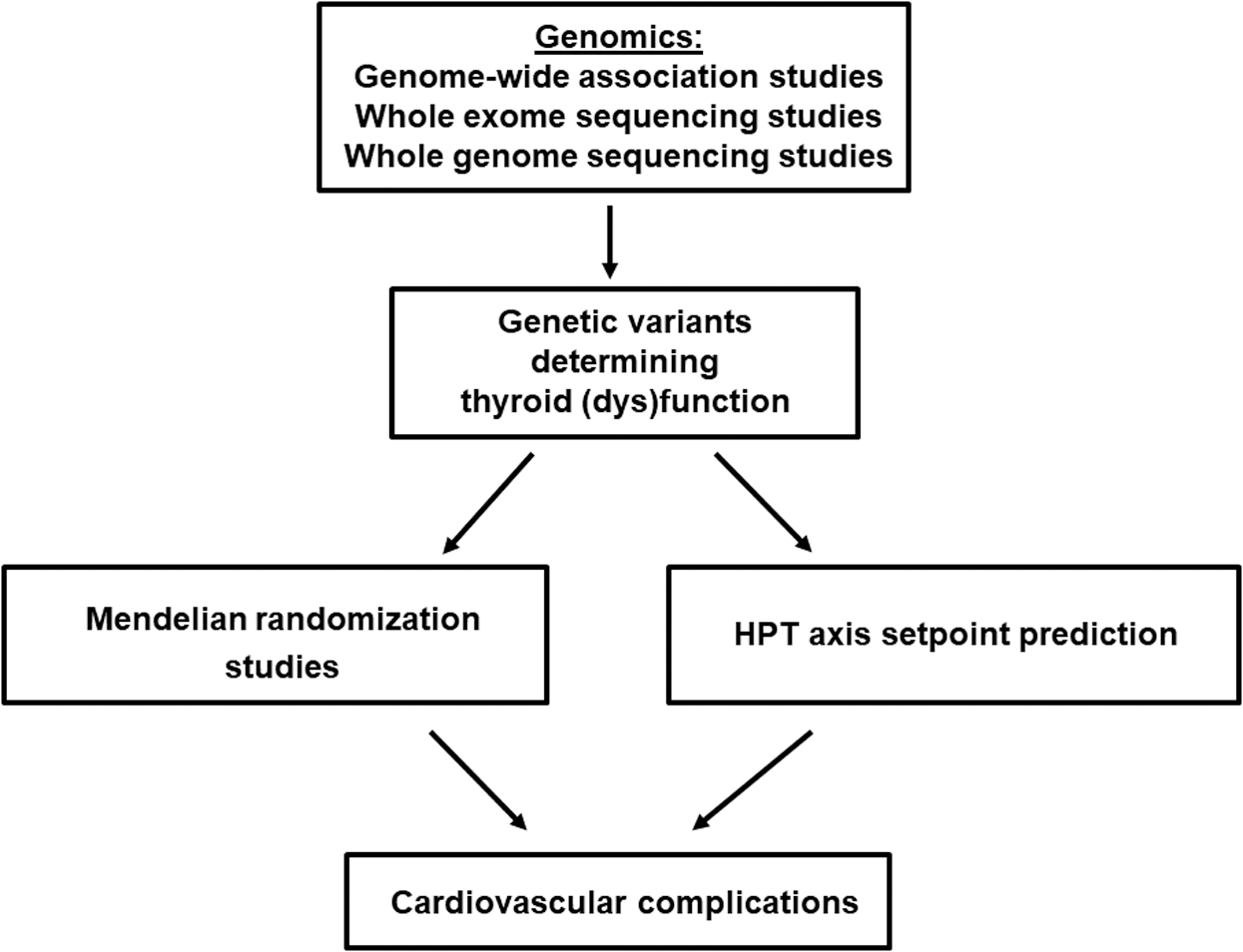
Role of big data in identifying novel genetic determinants of thyroid (dys)function. The identification of these markers is essential to perform reliable Mendelian randomization studies, which will clarify causality in the observed associations between thyroid (dys)function and cardiovascular complications. Furthermore, these genetic markers could play a role in hypothalamus–pituitary–thyroid (HPT) axis set-point prediction, which is essential to start personalizing the treatment of patients with thyroid diseases.
All current guidelines recommend TSH levels as the principal guide in the treatment of thyroid diseases because TSH is regarded as the most precise indicator of thyroid function (11). Whereas serum TSH reflects the thyroid status of the pituitary, other peripheral tissues might have a different thyroid status. This is nicely illustrated by patients with thyroid hormone resistance resulting from an inactivating mutation in thyroid hormone receptor (TR) β (38). Because TRβ2 is the TR in pituitary thyrotrophs, these patients have increased thyroid hormone levels with normal to high TSH levels. Although these TSH levels accurately reflect the thyroid status of TRβ-expressing tissues, they do not accurately reflect the thyroid status of TRα1-expressing tissues in the heart, which may be in a relatively hyperthyroid state, leading to tachycardia (38). Conversely, patients with TRα1 mutations experience bradycardia and low normal blood pressure (39). When aiming to prevent cardiovascular complications in the treatment of thyroid dysfunction, one would ideally guide treatment by a marker that specifically reflects the thyroid status of the cardiovascular tissues of interest.
Barriers and challenges
There are too few large population-based cohorts with data on thyroid function, omics, and cardiovascular end points. There has not been harmonization across cohorts of the assays used for thyroid function testing. In addition, a gap in most cohorts is the lack of measurement of T3 levels, and heterogeneity across cohorts in the use of total T3 and free T3 assays. In the search for the optimal clinical decision limit, sensitivity analyses in large well-phenotyped cohorts with available data on thyroid function, cardiovascular risk factors, and diseases are required. There may well be differences in decision limits between the various complications, which will require a consensus on the prioritization of the cardiovascular complications.
Besides thyroid dysfunction, other important risk factors such as age, sex, smoking, hypertension, and cholesterol levels determine a patient's cardiovascular risk profile. In further personalization of the management of thyroid diseases, the choice of when to treat should ideally be driven not only by thyroid hormone levels, but also by these other patient-specific characteristics. Finally, better biomarkers of the thyroid state in cardiovascular tissues are needed, as are hypothesis-free approaches to discover new pathways through which thyroid dysfunction leads to CVD.
Research opportunities
Collaboration among existing cohorts as well as harmonization of measurement variables such as T3 and free T4 will increase the availability of large data sets integrating data on CVDs, omics, and thyroid function. Such consortia can provide the unique framework necessary to perform adequately powered studies that can address many of the knowledge gaps described here. An initial assessment of the various potential cohorts with data available on thyroid function, genetics and omics, cardiovascular diagnoses, and medication use is required for determinations of analytical sample size. The majority of cohorts are lacking one of these essential data sources to participate in these analyses. Many study cohorts have stored blood samples, so it would be most efficient to start with the cohorts lacking thyroid function tests because these tests are relatively inexpensive and easy to determine. Investing in large sample sizes is also important to enable cross-ethnic and sex-specific analyses, as well as gene–environment interaction analyses, including smoking and diet (40). Comparison of the transcriptomics, proteomics, and metabolomics profiles with stratification based on the presence or absence of thyroid dysfunction and cardiovascular complications would be a hypothesis-free approach that could reveal new pathways underlying thyroid dysfunction and CVDs. Similar approaches have previously been proven to effectively identify new pathways involved in atherosclerosis, coronary artery disease, and diabetes mellitus (41
–43). The combination of the various omics, thyroid function, and CVD data is an excellent starting point in the search for proteins, microRNAs, or metabolites that specifically reflect the thyroid status of particular cardiovascular tissues. Because most of the available cohorts are population-based, findings first need to be confirmed in patient cohorts before they can be translated into clinical practice. Increasingly, registries, insurance databases, and research networks use integrated clinical data systems. Examples include the National Heart, Lung, and Blood Institute Biological Specimen and Data Repositories Information Coordinating Center (BioLINCC), Cardiovascular Research Network (
CARDIAC ELECTROPHYSIOLOGY AND ARRHYTHMIAS
In this section, we present a brief summary of laboratory-based and clinical studies of thyroid effects on cardiac electrophysiology and arrhythmias, with a separate discussion of amiodarone, followed by a brief discussion of barriers and opportunities and a listing of research opportunities.
Current state of the field
Laboratory-based studies of cardiac electrophysiology
Thyroid hormones strongly affect cardiac electrophysiology and rhythm through diverse effects on the activity of multiple ion channel subunits, transporters, and exchangers that ultimately regulate cellular excitability (Fig. 3) (44). Accordingly, changes in circulating thyroid hormone levels may alter cardiac excitability and conduction, resulting in heart block and bradyarrhythmias, as well as automatic, triggered, and reentrant supraventricular and ventricular tachyarrhythmias, most commonly atrial fibrillation. These effects depend on whether the thyroid hormone levels are increased or decreased, as explained below. A detailed understanding of the mechanisms through which altered thyroid hormone activity impacts cardiac electrophysiology is of substantial clinical and therapeutic importance.
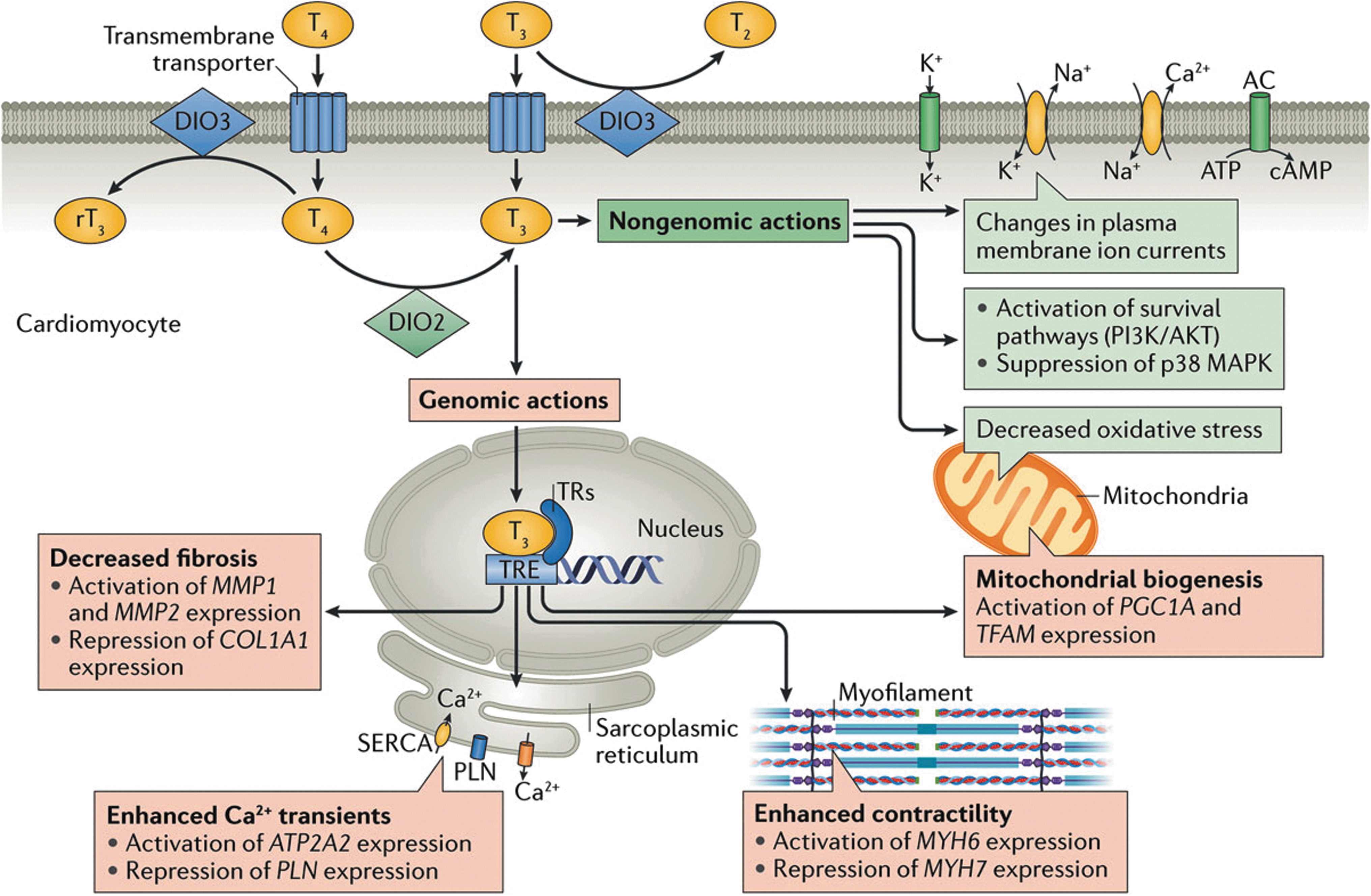
Effect of thyroid hormones on the cardiomyocyte via genomic and nongenomic actions from Reference (2). Reproduced with permission from Jabbar et al. (2). Copyright © 2017, Springer Nature Publishing AG. DIO2, type 2 iodothyronine deiodinase; DIO3, type 3 iodothyronine deiodinase; MAPK, mitogen-activated protein kinase; MYH6, myosin heavy chainα; MYH7, myosin heavy chainβ; PI3K, phosphoinositide 3 kinase; PLN, phospholamban; rT3, reverse T3; SERCA, sarco/endoplasmic reticulum Ca2+-ATPase; T2, diiodothyronine; T3, triiodothyronine; T4, thyroxine; TR, thyroid receptor; TRE, thyroid hormone response element.
Genomic effects
Initial molecular studies have identified widespread alterations in the “ion channel transcriptome” in response to altered thyroid hormone status (45). Analyses using cDNA microarrays have detected changes in the abundance of transcripts encoding multiple potassium, sodium, and calcium channel pore-forming and accessory subunits in response to experimental hyperthyroidism and hypothyroidism, usually associated with commensurate changes in ion channel current density as measured by patch-clamp recordings (45). These include prominent upregulation of the HCN2 pacemaker channel transcript in hyperthyroidism and, conversely, its suppression in hypothyroidism, consistent with changes in heart rate in these conditions (46). Thyroid hormone status also regulates gap junction channel expression and function (47). The hyperthyroid state can be associated with a decrease in ventricular fibrillation threshold (48), potentially a consequence of exaggerated catecholamine tone and/or altered cell–cell coupling.
The downstream components of the thyroid hormone signaling axis during heart formation and maturation are incompletely characterized. In situ hybridization studies of the developing murine heart suggest that TRα1 is enriched in the trabecular myocardium, whereas TRβ1 is only weakly expressed (49). These data are consistent with murine knockout studies, in which loss of function of TRα1 produces heart rate slowing and QRS and QT interval prolongation (50). However, the field could greatly benefit from a more precise examination of the individual cellular mechanisms that genomically mediate the thyroid hormone response.
Nongenomic effects
There is also some evidence that thyroid hormones influence cardiac excitability through TR-independent signaling mechanisms, potentially regulating several electrogenic proteins, including voltage-gated potassium channels, Na+/K+ ATPase, and Na+/Ca2+ ATPase activities (2). Certainly, evidence for nongenomic actions of thyroid hormone exists in a number of experimental models (51). However, additional information about these so-called nongenomic pathways is needed (52).
Clinical studies of arrhythmias
Clinically, patients with thyroid hormone excess have an increased risk of atrial fibrillation. The threshold of thyroid function at which that risk becomes clinically significant has been the subject of analyses of observational studies. In participants ≥60 years of age and older enrolled in the Framingham Heart Study, TSH ≤0.1 mIU/L was associated with a 3.3-fold increase in atrial fibrillation risk (53). A subsequent analysis of the Cardiovascular Health Study showed that there was a 2-fold increased risk of atrial fibrillation in individuals ≥65 years of age with a low TSH concentration (<0.45 mIU/L), even when free T4 concentrations were normal (subclinical hyperthyroidism) (54). There was a 1.85-fold increase in risk, even in those with TSH concentrations of 0.1–0.44 mIU/L. These findings have been confirmed in an individual patient data meta-analysis from the Thyroid Studies Collaboration (22).
Additional analyses have explored whether there is a gradient of risk for developing atrial fibrillation, even within the normal reference range of thyroid function tests. Data show increasing risk with decreasing TSH within the normal reference range in the Rotterdam Study (55) and with increasing free T4 within the reference range but not with concentrations of TSH or total T3 within their respective reference ranges in the Cardiovascular Health Study (56). This gradient of risk within the reference range was clinically significant in the older population (≥65 years of age) enrolled in the Cardiovascular Health Study, with an absolute risk difference of 11 per 1000 person years between the lowest and highest quartiles of free T4 (56). The association between free T4 within the reference range and atrial fibrillation was recently confirmed in a meta-analysis in the Thyroid Studies Collaboration (18).
The relative effects of using different thyroid hormone preparations—levothyroxine, L-triiodothyronine, and combinations of the two hormones (as with desiccated thyroid or synthetic combinations)—on arrhythmia risk have not been well characterized. Both endogenous T4 and levothyroxine have a 7-day half-life, whereas T3 and L-triiodothyronine have a 1-day half-life. T4 is converted to T3 through deiodination. However, levels of T4 and T3 differ between levothyroxine users and individuals in the euthyroid state at similar levels of TSH. Individuals with normal TSH levels who are taking levothyroxine therapy have higher serum free T4 concentrations while taking levothyroxine than when they were in the euthyroid state before a thyroidectomy (57) or compared with individuals in the euthyroid state not taking levothyroxine (58). In addition, levothyroxine users with exogenous subclinical hyperthyroidism have lower T3 levels than their nonuser counterparts with endogenous subclinical hyperthyroidism. These differences suggest that the risks derived from studies of endogenous subclinical hyperthyroidism may not apply to individuals with exogenous subclinical hyperthyroidism. Scottish registry data support an increased risk of arrhythmia in patients taking levothyroxine who have a TSH level ≤0.03 mIU/L, but no increase in risk when TSH lies between 0.04 and 0.4 mIU/L (59), although free T4 and T3 levels were not available in this study. There is concern that exogenous T3 taken in excessive amounts can precipitate arrhythmias, but the threshold at which this risk is increased, and the effects of single versus multiple daily doses, is not known.
Beyond its effects on the genesis of atrial arrhythmias, thyroid hormone can have a paradoxical effect on ventricular repolarization, as reflected in the corrected QT (QTc) interval. Hyperthyroidism has been associated with both QTc prolongation (60) and short QTc intervals (61), and hypothyroidism with QTc prolongation and a heightened risk for torsades de pointes (62). These observations may be attributed to the net effect in context of sympathetic tone, genetic and other medical characteristics of the patient, which determines whether and how a hypothyroid or hyperthyroid state might provoke either bradyarrhythmias or tachyarrhythmias. Thyroid hormonal imbalances, particularly hyperthyroidism, can also exaggerate the effects of catecholamines, enhancing the risk of arrhythmogenesis (63).
Amiodarone
Greater insight into some of the electrophysiological effects of thyroid hormone has been gleaned through understanding the actions of amiodarone. Amiodarone is a highly effective antiarrhythmic agent for both atrial and ventricular arrhythmias with complex electrophysiological effects, including calcium and β-adrenergic blocking properties, sodium and potassium channel blockade, and a structural similarity to T3. This similarity permits amiodarone to bind to thyroid receptors while being itself incapable of exerting any T3 agonist effects, and once bound, it may act as an antagonist (64). In addition, amiodarone inhibits the 5′-monodeiodinases type I and II enzymes responsible for conversion of T4 to T3, thereby blocking peripheral T3 production and raising circulating free T4 and lowering T3 concentrations. Amiodarone also inhibits entry of T4 into cells and the intracellular conversion of T4 to T3. Taken together, these actions can result in a functional hypothyroid condition at both systemic and cardiac tissue levels (65). Indeed, many of the therapeutic effects of amiodarone on cardiac automaticity, conduction, and tissue refractoriness mimic a state of physiological thyroid depletion, suggesting that the actions of the drug may be partly or even largely mediated through a thyroid mechanism. These insights afford an opportunity to elucidate arrhythmia mechanisms that might affect pharmacological approaches to arrhythmia management. In addition, amiodarone has been reported to modulate ion channel transcription through mechanisms not fully attributable to drug-induced local hypothyroidism (66). Given the widespread use of amiodarone for atrial fibrillation and other arrhythmias, a more complete understanding of these observations requires further investigation.
Although a highly effective antiarrhythmic agent, the long-term use of amiodarone may be abbreviated by toxicities to the thyroid gland and other organs (65). Depending on the underlying status of the thyroid gland, amiodarone may induce increased thyroid hormone production (type 1 amiodarone-induced thyrotoxicosis) or decreased thyroid hormone production (hypothyroidism) due to effects of the large amounts of iodine it contains (37.5% by weight; 75 mg per 200 mg tablet) on thyroid hormone production. Approximately 6 mg of iodine is released by the liver per 200 mg tablet. As a frame of reference, the recommended tolerable upper intake level in the United States is 1.1 mg of iodine per day. Amiodarone can also have a toxic effect on the thyroid gland, causing release of preformed thyroid hormone through thyroiditis (type 2 amiodarone-induced thyrotoxicosis).
Barriers and challenges
As discussed, significant knowledge gaps remain regarding the effects of thyroid hormone signaling within atrial and ventricular myocytes as well as specialized nodal and His-Purkinje cells. Experimental strategies to perform transcriptional profiling on each of these compartments are now well established (67), and extension of these methods to analyses of non-coding microRNAs and long non-coding RNAs are under development. In addition, the thyroid hormone sensitive gene regulatory networks that are modulated in various systemic disease states have been incompletely characterized.
Although a strong and consistent relationship between higher levels of thyroid hormone and incident atrial fibrillation has been shown, many questions about treatment remain. The target TSH or free T4 for preventing atrial fibrillation treatment and how this might vary by age, race/ethnicity, sex, and underlying cardiac status are not clear. No randomized clinical trials have tested whether treatment of mild hyperthyroidism prevents the new onset of atrial fibrillation or assists in the management of patients with preexisting atrial fibrillation. Nonetheless, several nonrandomized studies have shown a potential benefit of treating subclinical hyperthyroidism. These have shown improvements in heart rate, the frequency of atrial and ventricular premature complexes, left ventricular mass index, systemic vascular resistance, and exercise capacity after normalization of thyroid function (68 –70). Parallel knowledge gaps exist for the role of T3 in ventricular arrhythmias. There are as yet no efficient methods for identifying patients with thyroid dysfunction who are at particularly high risk for atrial or ventricular arrhythmias.
Approximately 10% of people ≥65 years of age take thyroid hormone replacement (71). Data indicate that excess levothyroxine replacement increases the risk of arrhythmias, but the threshold levels of TSH or free T4 at which this risk becomes clinically significant may not parallel those seen in patients with endogenous subclinical hyperthyroidism. Furthermore, there are patients who elect to take thyroid replacement preparations that contain T3. Because of the shorter half-life of T3, these preparations have more abrupt peak and trough effects. What represents a supraphysiological level of T3 in T3 replacement therapy and the impact that the resulting vacillations in T3 levels during the day have on the risk for tachyarrhythmias are unstudied.
The mechanism of the direct toxic effects of amiodarone on the thyroid or whether similar pathological processes might account for its other recognized toxicities requires further research. There are no known predictive characteristics to identify a high-risk phenotype for developing thyroid toxicity among patients in the euthyroid state who initiate amiodarone. The optimal frequency of thyroid testing in patients treated with amiodarone and the management of amiodarone-induced thyrotoxicosis and subclinical hypothyroidism also require refinement. Limited data exist on the dosing strategy to guide thyroid hormone replacement in patients taking amiodarone who develop hypothyroidism and on the thyroid function testing targets, along with the type and proper dose of thyroid hormone replacement in any patient with a history of arrhythmias. The two distinct forms of amiodarone-induced thyrotoxicosis have different treatments, and current methods to distinguish between them are imperfect, with some patients demonstrating overlap. Novel circulating markers and/or thyroid imaging techniques are needed for more accurate and prompt differential diagnosis.
Research opportunities
Specific research priorities defined by the Working Group related to the cardiac electrophysiological effects of thyroid hormone are summarized in Table 2.
Research Opportunities in Thyroid Hormones and Cardiac Electrophysiology
VASCULATURE AND ATHEROSCLEROSIS
In this section, we present a brief summary of thyroid effects on lipid metabolism and other cardiovascular risk factors and a review of clinical studies of thyroid status and cardiovascular events in patients without and with preexisting CVD, followed by a brief discussion of barriers and opportunities and a listing of research opportunities.
Current state of the field
Thyroid effects on lipid metabolism
There is considerable knowledge about the genomic mechanisms by which thyroid status affects lipid and lipoprotein metabolism. Although T3 induces HMG-CoA reductase, catalyzing the initiation of cholesterol biosynthesis, T3 also upregulates hepatic LDL receptor gene expression, mediated by TR-binding elements in the gene promoter region (72). Consequently, LDL cholesterol clearance is slowed in hypothyroidism (73). Thyroid status has also been shown to affect cholesterol 7α-hydroxylase activity, the first step in cholesterol degradation, and rates of fecal cholesterol and bile acid excretion (74,75). As a result of these actions, hypothyroidism has been associated with higher levels of LDL cholesterol and apolipoprotein B, as well as unfavorable changes in LDL particle number, size, and oxidation (76).
Thyroid hormone deficiency can increase circulating triglyceride concentrations through lesser activities of lipoprotein lipase, sterol-regulatory-element-binding protein-2, and apolipoprotein A1 (77). Thyroid hormone influences several aspects of HDL metabolism, upregulating hepatic lipase and cholesteryl ester transfer protein (78). In mice, thyroid status has been shown to alter reverse cholesterol transport by increasing levels of the hepatic HDL receptor SR-B1 (79). Finally, a decrease in thyroid hormone has been related to higher levels of atherogenic lipoprotein(a) (80).
In individuals with overt hypothyroidism, abnormal total and LDL cholesterol levels can be partially or completely normalized with thyroid hormone therapy (76,81). The favorable impact of thyroid hormone on the circulating lipid profile is further supported by the lower total and LDL cholesterol levels in endogenous and exogenous thyrotoxicosis (82) and the lowering of total and LDL cholesterol and triglyceride levels observed in clinical trials of TRβ agonists (83).
Increasing doses of the liver-selective TRβ agonist eprotirome were associated with LDL cholesterol reduction in a graded manner, with similar effects on apolipoprotein B, triglycerides, and lipoprotein(a) (84). The broad effects of eprotirome on lipids could derive from its specific pharmacodynamics or higher TRβ engagement in the liver compared with thyroxine.
The effect of subclinical hypothyroidism on lipids and lipoproteins is more controversial. A 2015 U.S. Preventive Services Task Force evidence review found that in eight trials of good or fair quality in which active levothyroxine treatment of subclinical hypothyroidism was compared with passive observation, mean serum total cholesterol reductions ranged from −28 to 0 mg/dL (85). In only three of these trials was total cholesterol lowering by levothyroxine supplementation statistically significant: −12, −28, and −12 mg/dL, respectively. Similarly, for serum LDL cholesterol, in eight trials of good or fair quality, differences between levothyroxine treatment and observation ranged from −22 to +2 mg/dL; and in only three trials was LDL cholesterol lowering by levothyroxine significantly greater: −8, −12, and −22 mg/dL, respectively. One recent study found that patients with subclinical hypothyroidism had higher small dense LDL particles—which are more atherogenic than larger less dense particles—only when serum TSH was greater than 10 mIU/L (86). For serum HDL cholesterol, the U.S. Preventive Services Task Force data review identified no trials finding a significant difference between the levothyroxine-treated and control groups. Finally, for serum triglycerides, in eight good and fair quality trials, differences between levothyroxine treatment and observation ranged from −32 to +11 mg/dL; and no trial found a significant difference between the levothyroxine-treated and control groups. In the Women's Health Study, higher TSH levels within the normal range and subclinical hypothyroidism were associated, in graded manner, with higher concentrations of small pattern B LDL particles and large VLDL particles (87).
Thyroid status and other cardiovascular risk factors
Approximately one-fourth of overtly hypothyroid patients have reversible, predominantly diastolic, hypertension. Indeed, blood pressure and TSH levels have been correlated, even within the reference range (88). However, the U.S. Preventive Services Task Force evidence review found no blood pressure–lowering effect resulting from levothyroxine treatment in two small trials (85).
Increases in plasma homocysteine levels have been reported in overt hypothyroidism and, in some studies, with subclinical hypothyroidism (89). Overt hypothyroidism has also been firmly associated with other atherosclerotic CVD (ASCVD) risk factors, including altered endothelial function and carotid intima-media thickness and higher uric acid and phosphate levels (90). In some studies, subclinical hypothyroidism has also been associated with other risk factors for ASCVD, including a hypercoagulable state, increased carotid intima-media thickness, decreased flow-mediated vasodilation and nitric oxide availability, and higher high-sensitivity C-reactive proteins levels (3).
Clinical studies of cardiovascular events
No clinical trials have been powered to examine the potential benefits of thyroid hormone supplementation on cardiovascular events in subclinical hypothyroidism; thus, events data are from population-based cohort studies and meta-analyses. An individual patient meta-analysis in the Thyroid Studies Collaboration found that subclinical hypothyroidism was associated with increased risk of CHD events in individuals with a serum TSH concentration ≥10 mIU/L and CHD mortality in individuals with a serum TSH concentration ≥7 mIU/L (16). However, the pathophysiological processes underlying the associations between thyroid dysfunction and CVD are incompletely understood. In studies investigating the relationship between thyroid dysfunction and cardiovascular end points, the associations remain statistically significant after adjustment for cardiovascular risk factors such as lipids and hypertension (20,22,23). This may indicate that thyroid dysfunction affects the risk of cardiovascular complications via other, nontraditional risk factor pathways.
In an observational study of patients in a UK research registry, patients 40 to 70 years of age with subclinical hypothyroidism who were treated with levothyroxine had fewer fatal and nonfatal cardiovascular events compared with nontreated individuals (91). However, no difference in these outcomes was seen between treated and untreated individuals >70 years of age.
Additional analyses have explored whether there is a gradient of risk for developing ASCVD, even within the normal reference range of thyroid function tests. In a meta-analysis from the Thyroid Studies Collaboration, there was no association between TSH within the reference range and cardiovascular events, with a trend toward increased risk at higher free T4 levels within the reference range (92). This trend was subsequently confirmed in the Rotterdam Study, with a higher risk of ASCVD events in participants with high-normal free T4 concentrations (93). This is consistent with findings from the Thyroid Studies Collaboration that demonstrated an increased risk of CHD events in participants with subclinical hyperthyroidism (22).
Clinical studies of thyroid hormone and preexisting CVD
The effects of correction of thyroid dysfunction in patients with preexisting CVD have not been studied. Low T3 syndrome and subclinical hypothyroidism are commonly observed after acute myocardial infarction and are associated with worse prognosis, though the role of thyroid hormone supplementation in mitigating ischemic–reperfusion injury in humans is not known (3).
Barriers and challenges
Few reports have examined the effects of thyroid hormone on atherosclerotic cardiovascular disease (ASCVD) in animal models. Limited data are available investigating variation in thyroid hormone action on the vasculature by sex or ethnicity. There are no randomized controlled trials of treatment of thyroid dysfunction with hard cardiovascular outcomes. Arguably, because there are other compelling reasons to treat patients with overt hypothyroidism, prospective studies of cardiovascular outcomes observing untreated individuals or comparing them with thyroid hormone–treated patients would be unethical. However, when to treat subclinical hypothyroidism, based on degree of thyroid dysfunction, age, and underlying cardiac status of the patient, remains unclear. A European trial investigating the impact of levothyroxine treatment in older individuals with persistent subclinical hypothyroidism changed its primary end point from ASCVD events to a symptom-based outcome because of high rates of reversion to euthyroidism and a delay in receiving the study drug (94). Additional studies are needed to identify alternative pathways underlying associations between mild thyroid dysfunction and CVD. There are no data on the effects of T3 therapy on vasculature or ASCVD. There are also no data on the optimal levothyroxine dosing strategy for patients with hypothyroidism who have existing ASCVD or to guide treatment of thyroid dysfunction in individuals after myocardial infarction. There have only been small and short trials studying the impact of thyroid hormone analogs in patients in the euthyroid state. Few observational studies have related the presence of thyroid autoimmunity to CVD, particularly on the basis of age, sex, and ethnicity.
Research opportunities
Specific research priorities identified by the Working Group for thyroid hormone and atherosclerotic vascular disease are provided in Table 3.
Research Opportunities in Thyroid Hormones and the Vasculature
HEART FAILURE AND THE MYOCARDIUM
In this section, we present a brief summary of the current state of the field, starting with the myocardial and hemodynamic effects of thyroid hormone and studies of thyroid dysfunction in new and established heart failure, followed by a brief discussion of barriers and opportunities and a listing of research opportunities.
Current state of the field
Myocardial and hemodynamic effects of thyroid hormone
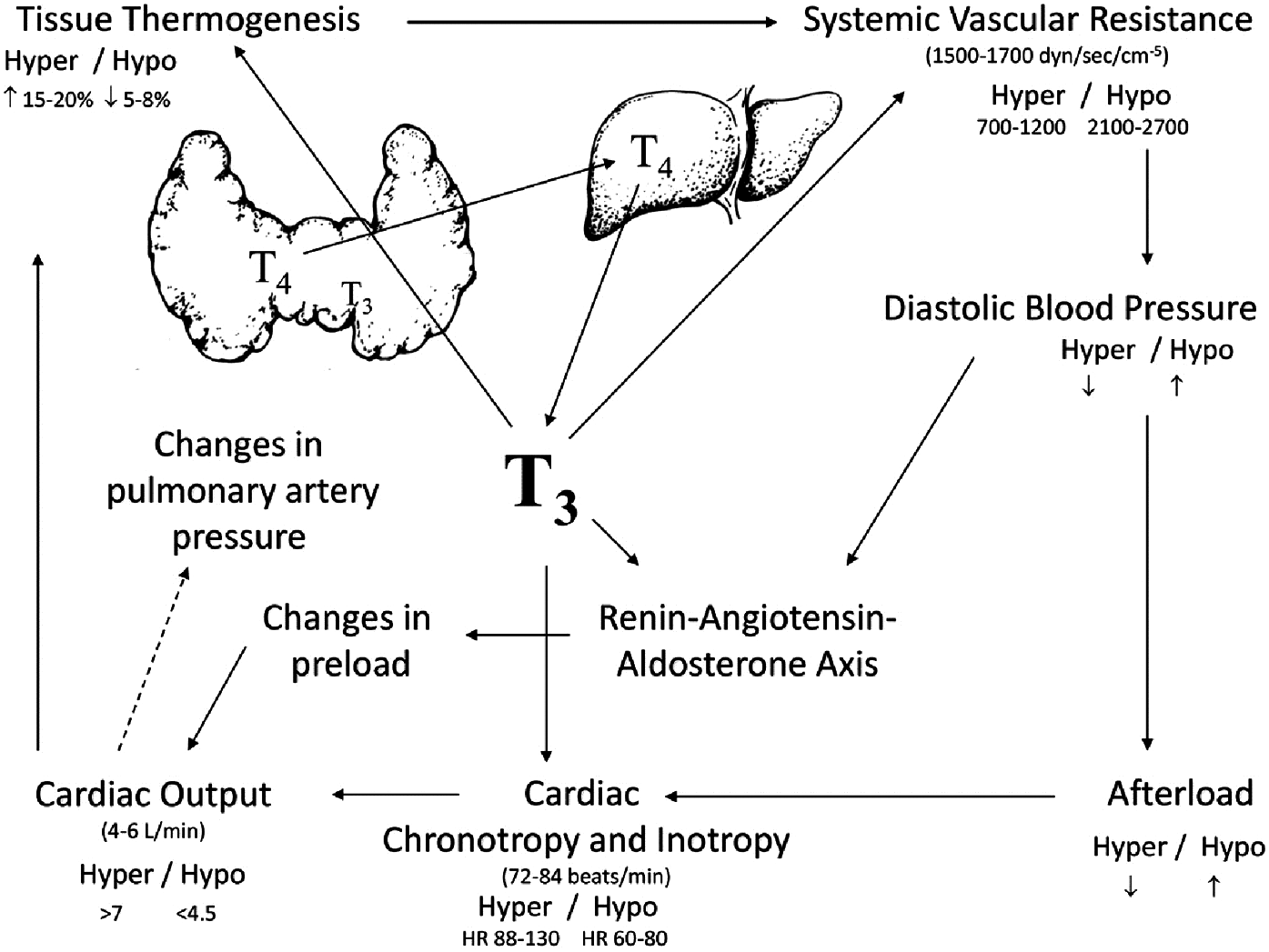
Thyroid hormones play an important physiological role in the regulation of myocardial function (Fig. 4) (2,95).
Genomic effects
T3 regulates the expression of genes encoding key components of the contractile apparatus, including upregulation of myosin heavy chain-α (MYH6) and downregulation of myosin heavy chain-β (MYH7), as well as key mediators of intracellular calcium handling, including sarcoplasmic/endoplasmic reticulum calcium ATPase 2 (SERCA2a) and its inhibitor phospholamban (PLN) (96,97). Increased levels of SERCA2a and decreased levels of phospholamban in response to thyroid hormone enhance ventricular relaxation in diastole by enhancing reuptake of calcium into the sarcoplasmic reticulum (98). Thyroid hormones also exert positive inotropic and chronotropic effects by enhancing expression of the β1-adrenergic receptor (99), although chronotropic effects of thyroid hormone do not require β-adrenergic signaling (100).
Nongenomic effects
Beyond these direct effects on the myocardium, thyroid hormone reduces systemic vascular resistance by enhancing production of endothelial nitric oxide and increasing calcium reuptake within the arterioles, thereby enhancing vascular smooth-muscle relaxation (101). Vasodilation is further enhanced by increased tissue metabolism and thermogenesis related to effects on mitochondrial function and by elaboration of vasodilatory peptides such as adrenomedullin and the natriuretic peptides, which are also in part regulated by thyroid hormone activity (102 –104). The increased cardiac output associated with thyroid hormone does not depend on a low systemic vascular resistance, but is associated with increases in blood volume and systemic venoconstriction that increase the pressure gradient for venous return (105). Secondary activation of the renin–angiotensin–aldosterone axis as a consequence of reduced mean arterial pressure leads to salt and water retention and to plasma volume expansion, which enhances ventricular preload and further augments cardiac output (106,107).
Thyroid dysfunction and the development of heart failure
The hyperdynamic circulation associated with hyperthyroidism, characterized by increased preload, high heart rate, and augmented left ventricular function, results in marked increases in cardiac output that may precipitate heart failure (108). Patients with hyperthyroidism also exhibit elevations in left ventricular mass compared with individuals with euthyroidism, which may represent a substrate for heart failure development (109). Although the precise mechanisms remain undetermined, thyroid hormone is known to participate in the pathogenesis of pulmonary vascular remodeling (110,111), and in population-based cohorts, there is a heightened prevalence of pulmonary hypertension in patients with hyperthyroidism (112). Incident atrial fibrillation in patients with overt hyperthyroidism and subclinical hyperthyroidism may also contribute to development of left ventricular dysfunction as a result of loss of the atrial contribution to end-diastolic volume and tachycardia-related effects on cardiac performance.
Patients with hypothyroidism exhibit impaired ventricular relaxation related to reductions in SERCA2a gene expression, increased expression of phospholamban, and associated reductions in intracellular calcium reuptake, as well as reductions in left ventricular stroke volume and cardiac index both at rest and during exercise (2). Untreated hypothyroidism can cause biventricular heart failure when profound and prolonged, but many of these physiological abnormalities can be reversed with thyroid hormone supplementation and restoration of a euthyroid status. These hemodynamic abnormalities likely contribute to the heightened risk of incident heart failure associated with subclinical hypothyroidism in individuals with serum TSH ≥10 mIU/L, as shown in an individual patient meta-analysis of the Thyroid Studies Collaboration (23).
Thyroid hormone in established heart failure
In patients with established heart failure, low T3 levels are common (as a result of impaired T4-to-T3 conversion), and the decline in serum T3 is proportional to the severity of left ventricular dysfunction (113). Reduced T3 in the presence of normal TSH and free T4 concentrations—the low T3 syndrome—is a powerful predictor of all-cause mortality in heart failure (114), even after adjustment for conventional predictors, including ejection fraction and natriuretic peptide levels (115). At the tissue level, diminished T3 bioavailability may be related to tissue hypoxia and inflammation, which conspire to reduce deiodinase activity in the cardiomyocyte, and also to an increase in DIO3 gene expression, which accelerates the degradation of T3 into inactive metabolites (116). Although in one small trial T3 replacement was associated with improvements in systolic and diastolic function and a reduction in neurohormonal activation (117), another study failed to confirm these benefits in patients with less severe heart failure (118). T3 is routinely administered to heart transplant donors and recipients to improve function, but no randomized studies support this practice (119). A prospective randomized trial of supplemental T3 in patients after coronary artery bypass grafting was negative (120). A multicenter clinical trial with a thyroid hormone analog, 3,5-diodothyroproprionic acid (DITPA), did not improve clinical outcomes in patients with heart failure but did increase cardiac index, decrease left ventricular size, and lower LDL cholesterol and body weight (121). In the Third National Health and Nutrition Examination Survey, in patients with preexisting heart failure, subclinical hypothyroidism was associated with increased mortality rates compared with control subjects with euthyroidism, especially in blacks (122). Clinical trials have not focused exclusively on this population.
Experimental data suggest that patients with heart failure may exhibit diminished responsiveness to thyroid hormone because of abnormalities in TR expression (123). Limited data in mouse hearts with ascending aortic constriction-induced cardiac hypertrophy and decreased cardiac contractile function show a marked decrease of TRα1and TRβ mRNA levels (124). Restoration of TR levels by adeno-associated virus (AAV) transgene expression significantly improves cardiac contractile function (124). In humans, a decrease in TRα1 has been reported in patients with cardiomyopathies (125).
Barriers and challenges
Thyroid hormones play a number of key roles in the regulation of cardiovascular performance. However, the possible contributions of thyroid dysregulation to the pathogenesis of heart failure remain insufficiently studied. Specifically, although overt hypothyroidism and hyperthyroidism are clear targets for therapy in patients with established heart failure, it remains unclear whether subclinical thyroid dysfunction requires similar attention. Similarly, the contributions of altered myocardial thyroid hormone actions in patients with euthyroidism with heart failure are still obscure. Although T3 bioavailability is thought to play a key role in modulating myocardial function, there is a lack of data regarding which specific myocardial effects of T3 play a significant role in development of human heart failure and might represent targets for therapy with T4, T3, or thyroid hormone analogs. Epidemiological data investigating subclinical thyroid dysfunction and preexisting heart failure are lacking, and the mechanism by which subclinical hypothyroidism and the low T3 syndrome enhance the risk of mortality in established heart failure patients remains to be studied.
Accurate quantification of tissue-level thyroid hormone action also remains a challenge. Although serum T3 levels can be measured easily, no methods are currently available for accurate quantification of tissue T3 levels, which may be more tightly correlated to myocardial function. Indeed, it is also not clear how thyroid hormone transporters, which are essential for T3 transport into cells, are regulated in the heart. There are also limited data on TR levels in animal models with heart failure and in patients with heart failure, and no data on contractile and electrophysiological effects of truncated TRα and TRβ proteins in cardiac myocytes.
The effects of thyroid hormone supplementation on heart failure prevention among those at risk and on cardiovascular outcomes in patients with established CVD also require further investigation. There are no prospective, randomized trials of thyroid hormone supplementation in patients with chronic heart failure with reduced ejection fraction or preserved ejection fraction and low serum T3 levels, and there is limited understanding of the optimal thresholds for therapy of thyroid hormone abnormalities in patients with established heart failure. Further data are needed on the role of thyroid abnormalities in key heart failure subsets such as those with heart failure and preserved ejection fraction or those with acute decompensated heart failure or cardiogenic shock.
Research opportunities
Specific research priorities on thyroid hormone effects on heart failure pathogenesis are summarized in Table 4.
Research Opportunities in Thyroid Hormones and the Myocardium
Conclusions and Recommendations
Based on discussion at the meeting, the Working Group defined three broad recommendations for research activity. The first set of recommendations focuses on basic biology. The second and third recommendations, with their focus on refining thresholds and testing treatment strategies in clinical trials, have more immediate translational potential.
Areas of particular interest for further research include the following: Defining the cellular signaling processes and gene expression variations by which thyroid hormone regulates electric conduction, contractility, and peripheral vascular function; Defining the cellular and molecular mechanisms relating thyroid hormone action to incident atrial and ventricular arrhythmias; Examining the role of thyroid hormone, its receptors, and cofactors in modulating myocardial systolic and diastolic function; Exploring the role of thyroid hormone in the setting of ischemia and its responsiveness to thyroid modulating agents; Exploring the role of thyroid hormone in modulation of endothelial function; Examining the mechanisms by which cardiac medications affect thyroid hormone production or action; and Developing novel serum and imaging biomarkers reflecting tissue thyroid hormone actions.
Areas of particular interest include the following: Identifying subgroups of individuals with preexisting CVD or at high risk for CVD who may benefit from manipulation of thyroid hormone status, including individuals in the euthyroid state. These studies would potentially support thresholds for treatment (based on specific levels of thyroid function tests) that vary by subgroup characteristics; Using modern approaches including genome-wide association studies, whole-exome/genome sequencing, epigenetics, mRNA expression, metabolomics, and other omics data to generate an HPT (hypothalamic–pituitary–thyroid) axis set-point prediction model to personalize treatment of thyroid dysfunction and test to what extent a deviation from the HPT axis set point is also important in determining a patient's cardiovascular risk; Recognizing the developmental and mechanistic differences among the newborn, pediatric, and adult populations; potential variations in key demographic subgroups, including age, sex, and race/ethnicity; and issues of health disparities and global health; and Leveraging the infrastructure from the two existing large consortia in this field (Thyroid Studies Collaboration and ThyroidOmics Consortium) to integrate data from existing large-scale cohorts or use existing registries.
Areas of particular interest for further research include the following: Designing studies, including small clinical trials designed to evaluate the feasibility and rationale for large intervention studies and test optimal strategies for management of subclinical hyperthyroidism, subclinical hypothyroidism, and low T3 syndrome in patients with and without preexisting CVD; these studies might test the treatment thresholds suggested by subgroup analyses of observational data; Designing studies, including small clinical trials, designed to justify large intervention studies of targeted thyromimetic analogs to treat dyslipidemia, heart failure, and peripheral vascular dysfunction; Incorporating appropriate examination of the HPT axis for new cardiovascular therapies for which preclinical data suggest an effect on the thyroid; Identifying appropriate surrogate (intermediate) end points for small clinical trials to help plan large intervention studies; and Establishing consortia to identify priority areas for therapeutic pharmacology studies and provide centralized protocol coordination, data management, and end point assessment for multicenter studies.
Disclaimer
The views expressed in this article are those of the authors and do not necessarily represent the views of the NHLBI; the National Institutes of Health; the U.S. Departments of Veterans Affairs, and Health and Human Services; or the U.S. government.
Footnotes
Author Disclosure Statement
Dr. Desai reports a research grant from Novartis and consulting fees from Novartis, Abbott, AstraZeneca, Boston Scientific, Bohringer-Ingelheim, Corvidia Therapeutics, Dalcor Pharma, Regeneron, Relypsa, and Signature Medical. Dr. Mora reports a research grant from Atherotech Diagnostics.
Appendix
Working Group Members:
Writing Group:
Anne R. Cappola, MD, ScM, Perelman School of Medicine at the University of Pennsylvania; Akshay S. Desai, MD, MPH, Brigham and Women's Hospital; Marco Medici, MD, PhD, MSc, Erasmus University Medical Center; Lawton Cooper, MD, MPH, National Heart, Lung, and Blood Institute, Debra Egan, MPH, National Center for Complementary and Integrative Health; George Sopko, MD, MPH, National Heart, Lung, and Blood Institute; Glenn I. Fishman, MD, NYU School of Medicine; Steven Goldman, MD, University of Arizona; David S. Cooper, MD, Johns Hopkins University School of Medicine; Samia Mora, MD, MHS; Brigham and Women's Hospital; Peter J. Kudenchuk, MD, University of Washington; Anthony N. Hollenberg, MD, Weill Cornell Medicine; Cheryl L. McDonald, MD, National Heart, Lung, and Blood Institute; Paul W. Ladenson, MD, Johns Hopkins University School of Medicine
Members:
Francesco S. Celi, MD, MHSc, Virginia Commonwealth University; Wolfgang Dillman, MD, University of California San Diego; Christina Ellervik, MD, PhD, DMSci, Boston Children's Hospital; A. Martin Gerdes, PhD, New York Institute of Technology College of Osteopathic Medicine; Carolyn Ho, MD, Brigham and Women's Hospital; Giorgio Iervasi, MD, Italian National Research Council; Amir Lerman, MD, Mayo Clinic; Ayako Makino, PhD, University of Arizona; Kaie Ojamaa, PhD, New York Institute of Technology College of Osteopathic Medicine; Robin Peeters, MD, PhD, Erasmus University Medical Center; Alessandro Pingitore, MD, PhD, Italian National Research Council; Salman Razvi, MD, Newcastle University; Ari J. Wassner, MD, Boston Children's Hospital