
Abstract
Background:
The differential diagnosis of thyroid nodules using fine-needle aspiration biopsy (FNAB) is challenging due to the inherent limitation of the cytology tests. The use of molecular markers has potential to complement the FNAB-based diagnosis and avoid unnecessary surgeries. In this study, we aimed to identify DNA methylation biomarkers and to develop a diagnostic tool useful for thyroid lesions.
Methods:
Genome-wide DNA methylation profiles (Illumina 450K) of papillary thyroid carcinoma (PTC = 60) and follicular thyroid carcinoma (FTC = 10) were compared with non-neoplastic thyroid tissue samples (NT = 50) and benign thyroid lesions (BTL = 17). The results were confirmed in publicly available databases from the Gene Expression Omnibus (GEO) and The Cancer Genome Atlas (TCGA) using the same DNA methylation platform. Two classifiers were trained to discriminate FTC and PTC from BTL. To increase the applicability of the method, six differentially methylated CpGs were selected and evaluated in 161 thyroid tumors and 69 BTL postsurgical specimens and 55 prospectively collected FNAB using bisulfite-pyrosequencing.
Results:
DNA methylation analysis revealed 2130 and 19 differentially methylated CpGs in PTC and FTC, respectively. The CpGs confirmed by GEO and TCGA databases showing high areas under the receiver operating characteristic curve in all sample sets were used to train our diagnostic classifier. The model based on six CpGs was able to differentiate benign from malignant thyroid lesions with 94.3% sensitivity and 82.4% specificity. A similar performance was found applying the algorithm to TCGA and GEO external data sets (91.3–97.4% sensitivity and 87.5% specificity). We successfully evaluated the classifiers using a bisulfite-pyrosequencing technique, achieving 90.7% sensitivity and 75.4% specificity in surgical specimens (five of six CpGs). The study comprising FNAB cytology materials corroborated the applicability and performance of the methodology, demonstrating 86.7% sensitivity and 89.5% specificity in confirmed malignant tumors, and 100% sensitivity and 89% specificity in cases with indeterminate cytology.
Conclusions:
A novel diagnostic tool with potential application in preoperative screening of thyroid nodules is reported here. The proposed protocol has the potential to avoid unnecessary thyroidectomies.
Introduction
Follicular cell-derived thyroid cancer (TC) is the most common endocrine malignancy, accounting for 90–95% of TC cases (1). Based on histology, these carcinomas are subdivided into well-differentiated carcinomas, including papillary thyroid carcinoma (PTC), follicular thyroid carcinoma (FTC), poorly differentiated thyroid carcinoma (PDTC), and anaplastic thyroid carcinoma (ATC) (1).
TC represents ∼5% of clinically palpable thyroid nodules (2). Fine-needle aspiration biopsy (FNAB) is the gold standard method for screening and diagnosis of thyroid nodules (3). Despite its high accuracy, nearly 17% of aspirations are classified as indeterminate cytology (4). According to The Bethesda System for Reporting Thyroid Cytopathology (5), cytologically indeterminate thyroid nodules comprise atypia of undetermined significance/follicular lesion of undetermined significance (Bethesda III), follicular neoplasms/suspicious for follicular neoplasm (Bethesda IV), and suspicious for malignancy (Bethesda V). Thyroidectomy is usually recommended for indeterminate or suspicious lesions; nevertheless, the majority will prove to be benign on the postsurgical evaluation (4,6). Therefore, these patients are submitted to an unnecessary surgery and will need thyroid hormone replacement throughout life (7,8).
Several molecular tests have been proposed aiming to improve the accuracy of indeterminate FNAB cytology and to avoid unnecessary and potentially harmful surgery for these patients (9 –16). The investigation for genomic aberrations in FNAB, including BRAF and RAS point mutations and PAX8/PPARγ and RET/PTC rearrangements, has been explored (17 –20). Recently, rare alterations described by targeted next-generation sequencing (16,19) have also been reported as being informative. Nevertheless, in many tumors, these alterations are not observed, resulting in an elevated number of false-negative results (14). However, they are useful to anticipate the surgical planning (“rule in” method). Additional methods show a greater ability to rule out the possibility of malignancy (“rule out” method) and could reduce unnecessary treatment (14). A “rule out” gene expression classifier based on 167 transcripts was tested in a multicentric study, including 265 FNAB with indeterminate cytology. A high sensitivity (92%), but a low specificity (52%), was found in the identification of suspicious nodules (9). Further studies confirmed the potential of this method (21,22); however, its clinical utility in avoiding surgeries remained unclear (23,24). The use of markers based on selected mRNAs (12) and miRNAs (13,25) has also been reported.
DNA methylation has been considered an attractive field for the development of clinical tools through the characterization of epigenetic modifications because of the higher stability of DNA compared with RNA (26 –28). Several genes have been described as differentially methylated in TC (29 –32), suggesting that these alterations could be useful to discriminate malignant from benign thyroid nodules. Herein, we investigated the use of DNA methylation markers as a potential diagnostic tool for TC.
Materials and Methods
Discovery and validation set of samples
The DNA methylation profile of 137 thyroid surgical samples from our previous study (31) was reassessed as a discovery set. These samples included 17 benign thyroid lesions (BTL; 8 follicular adenomas, 6 nodular goiters, and 3 cases of lymphocytic thyroiditis), 70 TC (60 PTC, 8 FTC, and 2 Hurthle cell carcinomas [HCC]), and 50 surrounding non-neoplastic thyroid tissues (NT) from PTC patients. To confirm the DNA methylation findings, 69 BTL (21 follicular adenomas, 47 nodular goiters, and 1 lymphocytic thyroiditis) and 161 TC (144 PTC, 12 FTC, 2 HCC, 1 PDTC, and 2 ATC) from patients who underwent surgery were included. Lymphocytic thyroiditis was obtained from patients submitted to thyroidectomy (mainly follicular adenomas associated with thyroiditis). The areas characterized by substitution of the thyroid parenchyma by lymphoid tissue were macrodissected. All BTL and 104 of 161 TC samples were not previously evaluated by microarray analysis (an independent set of samples). The institutional ethics committee (Protocols #475.385 and #2147.15) approved the study. The patients were advised of the procedures and authorized the research by signing an informed consent. Table 1 summarizes the clinical and histopathological data. DNA isolation, BRAF mutation genotyping, and bisulfite conversion procedure are described in detail in the Supplementary Data.
Clinicopathological Characteristics of the Patients Enrolled in the Study
Pathological examination or conclusive imaging examination (persistent disease included).
High serum thyroglobulin without imaging or pathological confirmation of recurrent disease.
ATC, anaplastic thyroid carcinoma; cN, no clinical evidence of cancer in the regional lymph nodes; FTC, follicular thyroid carcinoma; HCC, Hurthle cell carcinoma; Ni, not informed; PDTC, poorly differentiated thyroid carcinoma; pN0, no pathological evidence of cancer in regional lymph nodes; pN1, pathological confirmation of cancer in regional lymph nodes; PTC, papillary thyroid carcinoma.
Fine-needle aspiration biopsy
A total of 166 (FNAB) samples were prospectively collected (details in Supplementary Methods). The cytology of each nodule was evaluated and classified according to the Bethesda System (5). In cases of discrepancies, three specialized pathologists from the institution revised the samples and reached a final consensus.
Genome-wide DNA methylation analysis
The data previously generated by our group using the Infinium Methylation 450K assay (Illumina) (31) were re-evaluated under the perspective to identify epigenetic diagnostic markers (data available in Gene Expression Omnibus [GEO]; GSE97466). The sample groups (PTC, FTC, BTL, and NT) were compared using the Limma package® (33), and the differentially methylated CpG sites were identified with a Bonferroni-adjusted p-value <0.05 and a mean delta-beta (Δβ)> 0.2 or < −0.2. The quality control and processing of the microarray data are described in Supplementary Methods.
DNA methylation-based diagnostic classifier
The classifiers were constructed based on probes identified in the comparison between FTC or PTC versus NT and BTL samples (follicular or papillary-trained classifiers, respectively). This two-round classifier training was performed due to pronounced differences previously described in the DNA methylation profile of these two tumor types (31) and the higher incidence of PTC compared with FTC (1). Consequently, a straightforward “malignant against benign” training would prioritize the PTC classification, while FTC, which represents the most challenging entity in the cytology tests, would be misinterpreted. Probes presenting a high area under the receiver operating characteristic curve (AUC) were additionally filtered (AUC >0.95). The classifiers were trained by linear methods (support vector machine, diagonal linear discriminant analysis, and compound covariate predictor) using the BRB Array Tools v. 4.4.0 (Biometric Research Branch, National Cancer Institute). The performance was estimated by leave-one-out cross-validation (LOOCV).
Cross-study validation using external database
A cross-study validation was performed using genome-wide methylation data of thyroid tissues from GEO (GSE53051: 12 NT, 32 BTL, 18 FTC, and 20 PTC) and The Cancer Genome Atlas (TCGA) (515 PTC and 56 NT), retrieved from UCSC Xena. Both data sets were assessed in February 2018 and were used to confirm the differentially methylated CpGs in all comparisons (t-test p < 0.001 and FDR <5%) and to test the diagnostic classifiers.
Bisulfite-pyrosequencing analysis
The DNA methylation pattern of six selected CpGs (A: cg26307926; B: cg18395809; C: cg24240626; D: cg06533895; E: cg05471495; and F: cg25236791) was evaluated by bisulfite-pyrosequencing in the validation set of postsurgical samples (n = 230). Polymerase chain reaction (PCR) and sequencing primers were designed to evaluate the same CpGs identified by microarray analysis (Supplementary Table S1). After PCR amplification, a pyrosequencing reaction was carried out in the PSQ HS 96A Pyrosequencer (Qiagen, Valencia, CA) using PyroMark Gold Q96 reagents (Qiagen) (detailed in Supplementary Methods). The classifiers were retrained using the bisulfite-pyrosequencing values, adopting the same parameters used in the genome-wide methylation analysis, and evaluated in the FNAB samples (55 samples with sufficient DNA).
Statistical analysis
The results were statistically analyzed using SPSS v. 21.0 (Statistics Packet for Social Sciences, Chicago, IL) and GraphPad Prism software (v. 5.0; GraphPad Software, Inc., La Jolla, CA). Pearson's correlation test was applied to verify the concordance between microarray and bisulfite-pyrosequencing values. The biological groups were statistically compared by one-way analysis of variance (ANOVA, Tukey post hoc) and Student's t-test. The null hypothesis was rejected when the two-sided p-value was <0.05. The performance of the classifiers was estimated using sensitivity, specificity, positive predictive value (PPV), and negative predictive value (NPV) by MedCalc software v. 18.5 (MedCalc Software BVBA).
Results
Differentially methylated CpGs in PTC and FTC
We previously reported 3015 differentially methylated CpG probes in PTC (92% hypomethylated) and 5575 in FTC (74% hypermethylated) compared with NT (31). The reanalysis and new comparisons using the BTL samples revealed 4620 and 21 probes differentially methylated in PTC and FTC, respectively. Interestingly, most probes overlapped using both NT and BTL as references (PTC = 2130 and FTC = 19) (Supplementary Tables S2 and S3 and Supplementary Fig. S1).
The cross-study validation (GSE53051: 12 NT, 32 BTL, 18 FTC, and 20 PTC and TCGA: 515 PTC and 56 NT) comparing PTC versus NT and BTL confirmed 2333 (77.4%) and 3761 (81.4%) probes differentially methylated, respectively. A similar comparison of FTC versus NT and BTL revealed 3475 (62.3%) and 8 (38.1%) probes cross-validated by the GSE53051 data set, respectively (Supplementary Tables S2 and S3).
Detection of CpGs with high diagnostic potential
Cross-validated probes mapped in any genomic region (promoter, gene body, or intergenic region) presenting a high AUC in both internal and external data and capable to distinguish malignant from NT and BTL were further selected as candidate markers. Three CpGs (A: cg26307926; B: cg18395809; and C: cg24240626) (Fig. 1A) presenting AUC >0.95 and able to differentiate FTC from NT and BTL (AUC >0.75 for GSE53051 data set) were selected to compose the FTC-trained classifier. Similarly, three CpGs (D: cg06533895; E: cg05471495; and F: cg25236791) (Fig. 1B) (AUC >0.95 for GSE53051 data set) discriminated PTC samples from NT and BTL (PTC-trained classifier) (Table 2 and Supplementary Table S4).
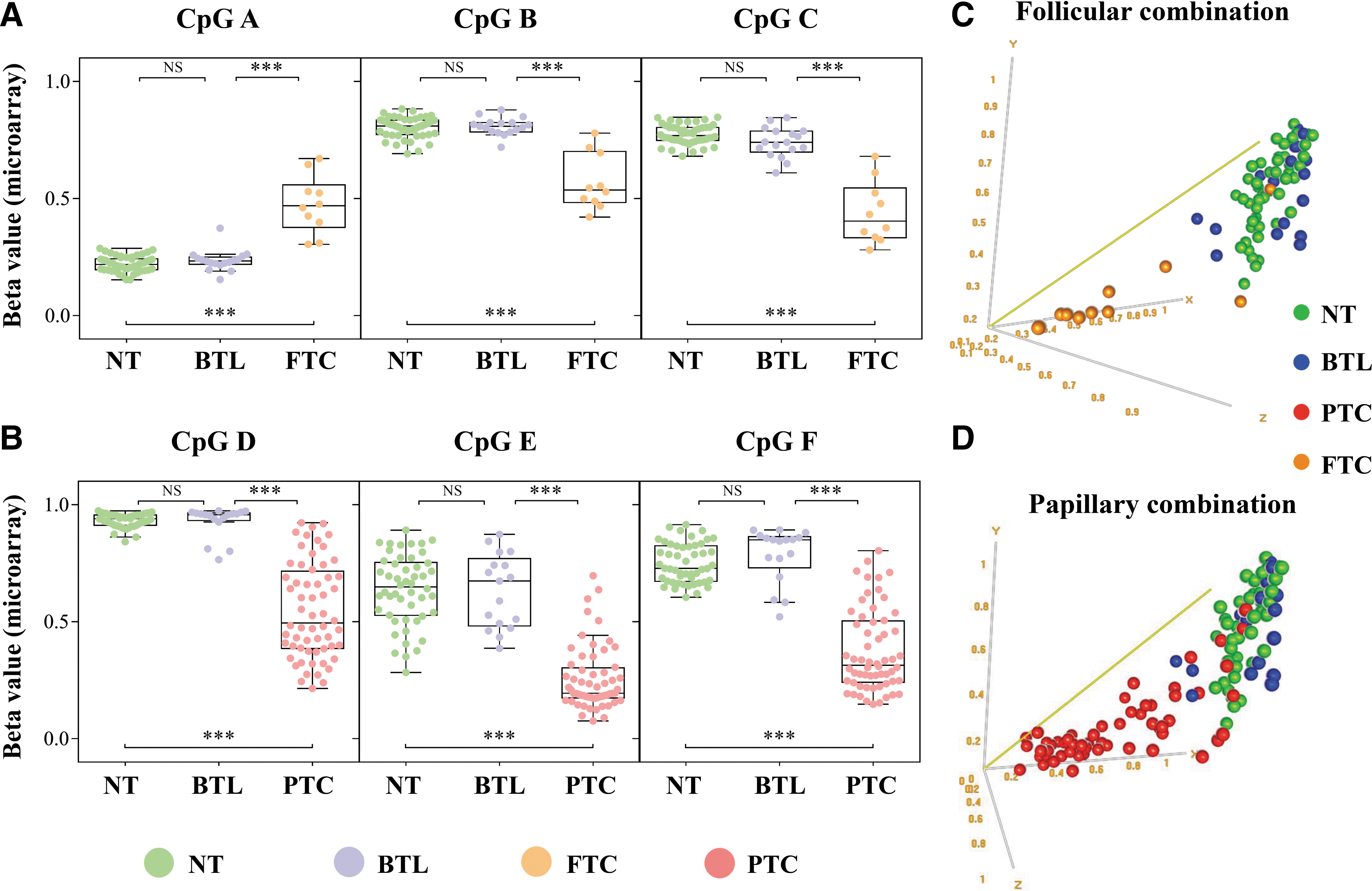
Differentially methylated probes in FTC and PTC with high diagnostic potential.
Description of the Probes Presenting High AUC and Selected to Train the Papillary Thyroid Carcinoma and Follicular Thyroid Carcinoma Classifiers
AUC, area under the ROC curve; BTL: benign thyroid lesions; CI, 95% confidence interval; NT, non-neoplastic thyroid tissue.
Training diagnostic classifiers for FTC and PTC using genome-wide DNA methylation data
Two distinct diagnostic classifiers were constructed to differentiate FTC from BTL (follicular-trained classifier) and PTC from BTL (papillary-trained classifier) (Fig. 1C, D). The best overall accuracy in LOOCV was achieved using the compound covariate predictor (CpG-A × 7.3 + CpG-B × −7.7 + CpG-C × −8, suspicious threshold > −7.5) and support vector machine (CpG-D × −1.8 + CpG-E × −2.2 + CpG-F × −1.3, suspicious threshold > −3.5) algorithms in follicular (96%)- and papillary (91%)-trained classifiers, respectively. The combination of both classifiers using microarray values was efficient to discriminate malignant from benign lesions with 94.3% sensitivity (66 of 70 malignant tumors), 82.4% specificity (14 of 17 BTL), 95.7% PPV, and 77.8% NPV. By applying the same model using the external data set (GSE53051), a sensitivity of 97.4% (37 of 38 malignant tumors), a specificity of 87.5% (28 of 32 BTL), a PPV of 90.2%, and an NPV of 96.6% was observed. Although DNA methylation data from BTL and FTC samples were not available in the TCGA database, 91.3% (470 of 515) of the PTCs were predicted as malignant tumors (Fig. 2). The individual prediction of the samples in the three microarray data sets is detailed in Supplementary Table S5. Three of the six selected CpGs showed differential methylation (ANOVA with a Tukey post hoc test p < 0.05) in both FTC and PTC (Supplementary Fig. S2), indicating that some predictions were not histological subtype specific. Nevertheless, our method was designed to distinguish malignant from benign thyroid diseases.

Flowchart showing the combined application of two classifiers and the performance indicating follow-up or surgery in all data sets (methylation microarray).
Confirmation of the CpG markers and adaptation of the methodology to bisulfite-pyrosequencing
To further confirm the findings and to readjust the methodology to a cost-effectiveness analysis for routine use, all six selected CpGs were assessed by bisulfite-pyrosequencing in PTC (n = 144), FTC (n = 14), three PDTC/ATC, and BTL (n = 69) samples obtained postsurgically. Significant positive correlations were observed between values obtained by microarray and pyrosequencing techniques (57 tumor samples evaluated by both procedures) (Fig. 3). By comparing the CpG methylation levels of PTC and FTC with BTL, the same pattern was observed (Fig. 4A).

Correlation between the methylation values obtained using microarray (beta values ranging from 0 to 1) and bisulfite pyrosequencing (% of methylation ranging from 0 to 100). r = Pearson correlation coefficient; p = p-value from Pearson correlation test; CpG-A: cg26307926; CpG-B: cg18395809; CpG-C: cg24240626; CpG-D: cg06533895; CpG-E: cg05471495; CpG-F: cg25236791.

Bisulfite pyrosequencing results in thyroid surgical and biopsy specimens. (
Diagnostic classifiers using bisulfite-pyrosequencing
The proportion of DNA methylated alleles generated by pyrosequencing was used for training new classifiers. The CpG-F was excluded, considering its modest correlation with the microarray results (r = 0.279) and the small difference between PTC and BTL (mean PTC = 85.8; mean BTL = 82.8). The strategy used for this approach was similar to the microarray analysis, adopting the compound covariate predictor (CpG-A × 2.4 + CpG-B × −3.6 + CpG-C × −5, suspicious threshold > −377.8) and support vector machine (CpG-D × −0.09 + CpG-E × −0.02, suspicious threshold > −8.47) methods in follicular (CpG-A, B, and C)- and papillary (CpG-D and E)-trained classifiers, respectively. The follicular-trained classifier allowed the correct identification of 57 of 69 BTL and 11 of 14 FTC (78.6% sensitivity and 82.6% specificity). Similarly, the papillary-trained classifier correctly categorized 62 of 69 BTL and 131 of 144 PTC (91.0% sensitivity and 89.9% specificity). Moreover, both models correctly classified very aggressive cases of TC as malignant lesions (one PDTC and two ATC). The combination of the classifiers presented a performance of 90.7% sensitivity (146 of 161 malignant tumors), 75.4% specificity (52 of 69 BTL), 89.6% PPV, and 77.6% NPV (Fig. 4B). The classification of each sample is detailed in Supplementary Table S6.
Curiously, among the 15 tumors incorrectly classified as benign, no evidence of recurrence (mean follow-up of 107 months, varying from 43 to 174 months) or extrathyroidal extension was found (frequency of 38.5% in the entire cohort of cases), and only one patient (6.7%) presented cervical lymph node involvement at diagnosis (observed in 34.1% of the cases). In our PTC samples, a BRAF mutation was detected in 68.8% (99/144) of cases and the sensitivity of the classifier could be increased to 91.2% with the addition of BRAF genotyping (two misclassified PTC harbored BRAFV600E ).
Bisulfite-pyrosequencing analysis in prospectively collected FNAB
The bisulfite-pyrosequencing assays were successfully performed in 55 of 166 samples obtained from the leftover cells inside the needles used for FNAB, which were prospectively collected (>10 ng of total DNA yield and high quality of the pyrosequencing). Eight indeterminate samples were further excluded, either because there was no indication for (patient is presently followed by observation) or persistent inconclusive results were obtained.
Application of the follicular and papillary diagnostic model resulted in a correct classification of 86.7% (13 of 15) of the conclusive malignant cases (12 Bethesda VI and 3 PTC-positive lymph nodes with postsurgical confirmation) and 89.5% (17 of 19) of the conclusive benign cytology cases (all Bethesda II without postsurgical confirmation). In the indeterminate group of samples, seven FNAB cytologies (postsurgically confirmed as PTC) were predicted as malignant (100% of sensitivity) and five of six postsurgically confirmed benign diseases were predicted as benign (83.3% of specificity) (Fig. 4C). Two misclassified Bethesda VI tumors were BRAF wild-type nonencapsulated classic papillary thyroid microcarcinomas. Therefore, BRAF genotyping was not able to increase the sensitivity of the test. The results obtained for each sample are detailed in Supplementary Table S7.
Discussion
DNA-based molecular biomarkers are very attractive due to the robustness and stability of these molecules (34). The first studies in this field evaluated the most common mutations and rearrangements that could identify ∼60% of the TC with indeterminate results based on cytological diagnosis (17,35). The addition of several other mutations and gene fusions using next-generation sequencing panels brought an increase in the sensitivity up to 91% (19). However, BTL also often harbor driver alterations, mainly point mutations in the RAS genes (36,37). Moreover, a considerable fraction of TC lack the most common genetic alterations (1,29), leading to a loss of sensitivity.
Using a comprehensive genome-wide methylation profiling technology, capable of detecting DNA methylation patterns at a single-site resolution, we determined the methylation alterations in the main subtypes of follicular-derived thyroid tumors. These alterations were used to develop two classifiers, which in combination allowed the discrimination of malignant from benign lesions with high sensitivity and specificity (94.3% and 82.4%, respectively). An external set of samples was used to guide the selection of the most promising individual markers, rather than validating the classifiers (which presented comparable classification performance).
Despite the large number of probes and high reproducibility noted in the cross-validation of PTC against BTL comparison (3761 cross-validated probes), a limited number of probes were observed in the comparison between FTC and BTL (8 cross-validated probes). In fact, the diagnosis of FTC is a challenge, and the agreement among experienced pathologists is low (∼57%) (38). As we previously reported, the methylation profiles of FTC and follicular adenomas can also be very alike (31).
To identify FTC in FNAB, three CpG sites were incorporated in our method, including a hypermethylated CpG in an intergenic region of chromosome 2 (CpG-A) and a hypomethylated CpG in the gene body and the 5′UTR of PTPRN2 (CpG-B) and REG3A (CpG-C), respectively. Curiously, hypermethylation of PTPRN2 was described as a putative diagnostic biomarker in lung cancer (39,40), and in cell-free circulating DNA from patients with this tumor type (41). In addition, increased expression of REG3A was associated with proliferation, migration, and invasion of gastric (42) and colorectal tumor cells (43). The REG3A promoter DNA methylation appears to be an important regulatory mechanism of its expression (44).
Since PTC is the most common thyroid malignancy, we included three frequently hypomethylated CpGs in PTC mapped in the 5′UTR of DCUN1D3 (CpG-D), the intergenic region of chromosome 2 (CpG-E) and in the gene body of WDR82 (CpG-F) in the method. However, the discrimination power of WDR82 was not reproducible in the pyrosequencing analysis.
The potential of DNA methylation markers in the differential diagnosis of thyroid lesions has been explored in the literature. An abnormal DNA methylation pattern of RARB2 was reported as marker to identify anaplastic carcinomas (45), while hypomethylation in LGALS3 and hypermethylation in CDKN2A, TSHR, and RASSF1A were able to distinguish PTC from non-neoplastic thyroid tissues (46,47). The DNA methylation levels of CALCA, TIMP3, DAPK1, CDH1, and RARB2 were tested in circulating DNA of 53 patients submitted to FNAB (with surgical results in 31 PTC, 7 FTC, and 15 BTL). The authors reported 77% of accuracy, with a high PPV (96%) and a low NPV (60%) in discriminating malignant from benign disease (26). A similar panel was tested in FNAB from PTC patients (including RASSF1A instead of CDH1) with the addition of BRAF genotyping (27). The authors found 85.7% of accuracy in a training group (n = 79) and 79.7% in the validation set of cases (n = 38). The 5-hydroxymethylation analysis of cell-free circulating DNA was suggested as a useful early diagnostic strategy for colorectal, stomach, pancreas, liver, and TC (28). Recently, Yim et al. described an epigenetic signature of 137 CpGs resembling benign lesions (n = 28) and 236 CpGs for PTC (n = 39) (48). The authors developed and tested a model using PTC samples (n = 65) achieving 100% sensitivity and 97% specificity. The follicular variants of PTC and FTC that represent the main diagnostic difficulties (38) were not included in most of these studies, which could overestimate the performance of the tests.
The use of single markers could generate false-positive and false-negative results, which are inappropriate for clinical application purposes. However, the inclusion of many markers in a “multigenic” system could confer an overfitting in the model (49). When the degrees of freedom in the parameter selection exceed the data information index, the model becomes arbitrary and hardly reproducible. To overcome this obstacle, the addition of an increased number of samples is required (49,50). Since only a small fraction of the biological material is usually available in clinical routine, an effort was made to include few CpG markers and consequently enhancing the viability of the methylation test. Considering that DNA methylation microarrays are not suitable in the clinical setting, we adapted our methodology using bisulfite-pyrosequencing, a well-established procedure already used for clinical purposes. Consequently, a low per sample cost and turnaround time of two to three days from biopsy to the final molecular report (sample processing, DNA isolation, bisulfite conversion, PCR, pyrosequencing, and analysis) would be conceivable. Highly significant correlations were observed between the cases evaluated by both microarray and bisulfite-pyrosequencing (n = 57) for five of the six assayed CpGs (r > 0.5 and p < 0.001). The CpG-F had a low correlation coefficient (r = 0.28, p = 0.047) and was excluded from subsequent analysis. Optimal concordance between the microarray and pyrosequencing was previously reported (51).
We obtained 90.7% sensitivity and 75.4% specificity using 230 thyroid samples, including 173 microarray-independent cases, evaluated using the bisulfite-pyrosequencing protocol. The values of 89.6% PPV and 77.6% NPV obtained are greatly influenced by the prevalence of malignancy, as previously reported (52). Therefore, the overall accuracy of our data was superior to other molecular tools used in clinical settings (53), such as the Afirma gene expression classifier (GEC, Veracyte, Inc., San Francisco, CA), with the advantage of using DNA at a very low concentration (ranging from 10 to 125 ng of DNA) adding to the lower cost of the procedure.
Curiously, most of the misclassified tumors by our approach presented indolent behavior (no extrathyroidal extension, low frequency of node metastasis, and no recurrence in the follow-up). In fact, the most common types of TC are associated with an excellent prognosis and avoiding overtreatment is currently one of the main problems in thyroidology (1).
In our study, FTC and the follicular variant of PTC samples were included in both genome-wide DNA methylation and specific CpG-site analysis. The pyrosequencing assays of surgical specimens correctly classified 32 of 40 (80%) FTC/HCC and follicular variants of PTC. Importantly, three very aggressive thyroid tumors (one PDTC and two ATC) were included in the analysis, and all were scored to be malignant in both the PTC- and FTC-trained classifiers. The tumors defined as benign by the test were 2 minimally invasive FTC (2 of 14 FTC/HCC), 6 PTC follicular variants (6 of 26), 5 PTC classic variants (5 of 108), and 2 rare PTC variants (oncocytic and diffuse sclerosing) (2 of 10). By combining the analysis with BRAF genotyping, two of these tumors, one classic and one PTC follicular variant, could be identified as malignant. Although just a slight improvement in sensitivity was observed (from 90.7% to 91.2%), the inclusion of BRAF mutational analysis before bisulfite conversion is of interest given the high predictive value of the BRAF mutation. This alteration can be easily evaluated even with very small amounts of DNA (54) and occurs with high frequency in PTC, PDTC, and ATC (1). Among the BTL classified as malignant, 6 follicular adenomas (of 17 tested) and 11 nodular goiters (of 47 tested) were included. We previously reported similar methylation characteristics of BTL (mainly follicular adenomas) with minimally invasive FTC, which tend to group together in an unsupervised clustering analysis (31).
A preliminary study was implemented with 166 nodules surveyed by FNAB cytology to further certify the feasibility of the CpG methylation-based tool. However, only 63 samples were considered suitable for pyrosequencing analysis (>10 ng DNA) and 55 had high-quality results. One hundred three samples were excluded due to insufficient DNA yields from the remaining cells in the needle (less than 10 ng). In the practical scenario, an additional pass dedicated to DNA extraction appears necessary for reliable molecular assays. For ethical reasons, additional biopsies were not collected exclusively for the assays. Despite the small sample size, the method was able to correctly classify 20 of 22 malignant cytology samples (malignancy confirmed postsurgically). The two misclassified samples were BRAFV600E -negative indolent tumors (pT1N0M0), similar to the analysis performed on surgical samples. None of the FNAB samples investigated had a postsurgical histology of noninvasive follicular thyroid neoplasm with papillary-like nuclear feature.
Similar good performance was observed in Bethesda II and indeterminate cytology cases reported as benign postsurgically (or a new FNAB in the same nodule with Bethesda II), properly suggesting clinical follow-up in 22 of 25 samples. Curiously, one Bethesda II sample (FNAB6) considered malignant by the test was from a patient harboring a PTC in the opposite thyroid lobule. The second Bethesda II sample (FNAB15) was under observation and the indeterminate benign lesion (FNAB66) had a follicular adenoma confirmed after lobectomy.
Taken together, we report a tool based on DNA methylation analysis presenting high accuracy and applicability in TC diagnosis. Five CpGs were selected from genome-wide DNA methylation profiles obtained from internal and external data set analysis. Bisulfite-pyrosequencing, a well-established approach, was used to investigate these five CpGs showing 90.7% sensitivity and 75.4% specificity in surgically resected thyroid samples. The analysis conducted in FNAB samples demonstrates the applicability and good performance, revealing 90.9% sensitivity and 88.5% specificity (100% of sensitivity and 89% of specificity in cases with indeterminate cytology). A main limitation of our study is the limited number of FNAB cases (n = 55), since the DNA yields from the needle washouts were insufficient in many samples to perform the experiments. The effectiveness of this novel diagnostic tool should be further verified in a prospective multicenter study.
Footnotes
Acknowledgments
The authors thank the A.C. Camargo Cancer Center Biobank for providing and processing the surgically resected samples, Mrs. Graziela Machado Gruner Turco Spilborghs for assisting in the prospective collection of FNAB, and Dr. Patricia Maria Peresi for her contribution with the selection of FNAB cases.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported by grants from the São Paulo Research Foundation (FAPESP 2015/20748-5 and 2015/17707-5) and CNPq (140819/2011-8).
Supplementary Material
Supplementary Data
Supplementary Figure S1
Supplementary Figure S2
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
Supplementary Table S5
Supplementary Table S6
Supplementary Table S7